
My previous post about information jumps, based on the example of the Prickle 1 protein, has generated a very interesting discussion, still ongoing. I add here some more thoughts about an aspect which has not been really analyzed in the first post, and which can probably contribute to the discussion. I will give here only a very quick summary of the basic issue, inviting all those interested to check my first post: Homologies, differences and information jumps and the following discussion, amounting at present at more than 500 posts. So:
- Prickle 1 is an interesting protein, with rich functional properties, in general still not well understood.
- With reference to the human form, I have identified two different parts in the protein. A first part with 4 identifiable domains, and a second part with no identifiable domain. In the following discussion, I will continue to conventionally refer to those two sequences as “the red sequence” and “the blue sequence”, according to the colors by which I have marked them in the first post.
- The red sequence is present in all eukaryotes, starting from fungi, and shows increasing levels of homology to the human form.
- The blue sequence, as derived from the human form, is restricted to vertebrates. It is practically absent in all other metazoa.
- Just from the beginning of the vertebrate “tree”, the blue sequence shows a very high homology to the human form: about 600 bits of conserved information sequence between sharks and humans. IOWs, the blue sequence is highly conserved in vertebrates, just form the beginning.
- For all those reasons, I assume that the blue sequence is functional in vertebrates, and that its rather sudden appearance at the root of the vertebrate tree in a very specific form is a good example of an information jump in natural history.
OK. This is more or less the essence of the first post. The following discussion has touched many aspects, but I will not mention them all here, because I am confident that they will surface again in the discussion about this post. Now, I will try to sum up in advance what I am going to discuss in this second post, and then show the facts that support my ideas. So:
- In the first post, I have focused on the human form of the protein, and used its two sequences to measure different levels of homology in metazoa.
- The blue sequence in humans has been found to be highly conserved in vertebrates (and therefore almost certainly functional), and amazingly restricted to them.
- But what about other metazoa? The important point is: there is always a “blue sequence” in the Prickle 1 protein, in all taxa. But it is completely different from the blue sequence in vertebrates.
- The main point of this post is to demonstrate that the blue sequence in Prickle 1 is a good example of a functional sequence which is highly taxonomically restricted.
OK. I hope this is clear enough.
Just to start the discussion, let’s look at the Prickle 1 protein in tunicata, which are (with cephalochordata) the nearest neighbour of vertebrata: they are, indeed, chordates, but not vertebrates. If we compare the human Prickle 1 to the available data for tunicata, we find a few hits, all of which are relative to the red sequence. The human blue sequence, as already said. has no significant homology in tunicata. Now, let’s consider the hit with the human Prickle 1 protein in Ciona intestinalis, the reference organism for Tunicata. We have 456 bits of homology, a very good value, and the alignment is exclusively relative to the red sequence, IOWs, the domain sequence.
But what about the blue sequence in Ciona intestinalis? Is there a blue sequence, at all?
The answer is: yes, definitely. The Prickle 1 protein in Ciona intestinalis is 1066 AAs long, indeed a little bit longer than the human form (which is 831). And here too, like in the human form, the domain part is confined to the first part of the molecule. IOWs, we have a blue sequence which is about 712 AAs long, and is found after the conserved red sequence. Of course, that blue sequence has no homology to the blue sequence in vertebrates. We already know that, but just to check I blasted the two blue sequences one vs the other. The result? 20 bits, expect 0.30. IOWs, absolutely no homology.
So, the Prickle 1 protein in Ciona has its blue sequence, which is completely different from the one in vertebrates.
First question: is it taxonomically restricted?
You bet! If we blast that 712 AAs sequence against all existing organisms, including prokaryotes, we get only 3 hits: one is with itself, Prickle 1 in Ciona: 1447 bits, the identity value, IOWs, the maximum informational value of the sequence. The second is with Prickle 2 in Ciona, 1255 bits, an almost identity. The third is a partial but significant hit (107 bits, 2e-20) with a not well defined prickle protein in Molgula tectiformis, another organism in Tunicata.
That is certainly taxonomically restricted at its best!
What about Cephalochordata? Blasting human Prickle 1, we find only one protein in lancelet, “hypothetical protein BRAFLDRAFT_121177”. It seems to be a Prickle 1 protein, with the usual 497 bits of homology confined to the red sequence. The whole protein is 842 AAs. That means that here too there is a blue sequence after the domain part. I have blasted this sequence (366 AAs): only one significant hit, the identity hit with itself (751 bits). So, this sequence too is absolutely taxonomically restricted. Let’s go back in time. Human Prickle 1 against Cnidaria, the best hit is a protein in Hydra vulgaris, named “prickle-like protein 3”. Again, a good homology for the domain sequence: 423 bits (expect 4e-135). Again, the protein is much longer (724 AAs), and there is a blue sequence after the red one. Again, I have blasted it (359 AAs) against all organisms: 2 hits, the identiy (729 bits) and a good 297 bits homology with another cnidarian protein, “LIM-domain protein prickle” in Clytia hemisphaerica.
Taxonomically restricted? Yes.
OK, I think you get the idea. For the convenience of the readers, I have summed up a few results which I have obtained by following the same procedure as in the previous examples. Identifying human Prickle 1 homologues in some group of organisms, identifying the associated blue sequence in a representative of the class, then blasting that sequence against all living organisms, to evaluate how restricted it is. Here are some results:
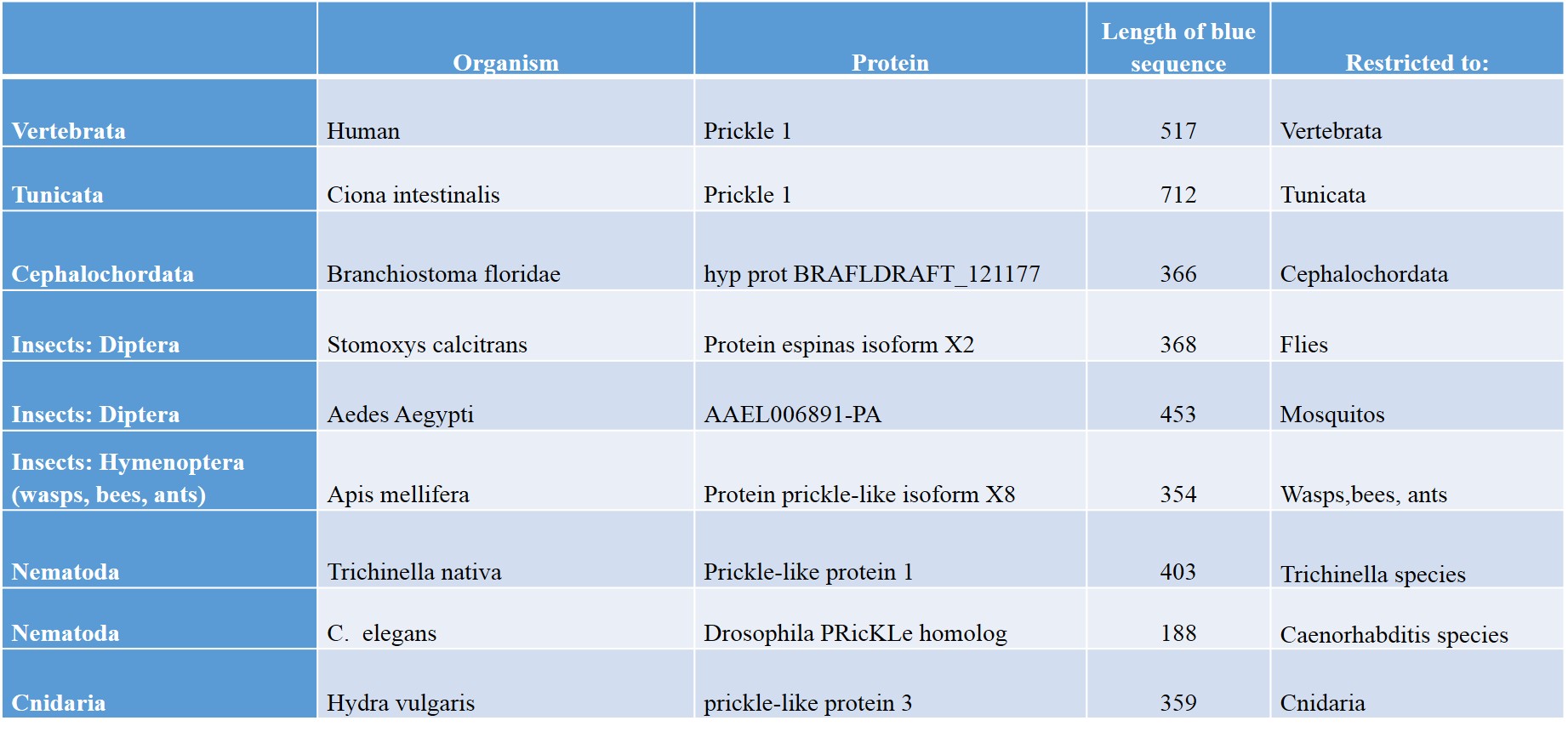
As everyone can see, different groups of organisms have different blue sequences, and those sequences are usually very much taxonomically restricted. For example, in Nematoda we have two completely different blue sequences in Trichinella species and in Caenorhabditis species.
Functional sequences?
OK, now the obvious question is: are these blue sequences functional? I strongly believe that we have reasons to think they are.
First of all, we have the example of vertebrates. The great number of sequenced genomes in vertebrates allows us to be sure that the blue part of the vertebrate sequence of Prickle 1 is highly conserved in practically all vertebrates, starting from cartilaginous fishes. That means high conservation for 400+ million years, an undeniable marker of negative/purifying selection and therefore of functional constraints. Those points have been discussed in depth in the previous posts and in the following discussion. Nobody has seriously denied that the blue sequence in the vertebrate Prickle 1 protein is functional.
What about other groups of organisms? Sometimes, the sequences are really very restricted, like in the case of Ciona intestinalis. Of course, in many cases that is also due to less biodiversity in the group, and/or lower number of sequenced genomes. So, it is much more difficult to ascertain how conserved each blue sequence is. However, let’s consider the following points:
- In most metazoa, with the exclusion of the simplest ones, and of unicellular eukaryotes, we find easily clear homologues of the Prickle 1 protein as we know it in vertebrates and humans. Of course, it’s the red part of the molecule, the domain part, which guides the identification, because of its good conservation in all those organisms.
- The rest of the molecule is different in different groups. Completely different, in most cases.
- However, in practically all homologues of Prickle 1, there is always an accessory sequence, usually after the domain part (there are some exceptions, where it precedes the domain part).
- It seems quite reasonable that all, or almost all, these homologues of Prickle 1 in various species are functional, even if the function could be different from species to species, at least for the regulatory effects of the protein network where the Prickle 1 is included. It is also reasonable that the domain part has similar functions in different contexts, while the accessory blue sequence is more linked to the local context.
This is, IMO, a very reasonable explanation of what we observe. Can we support these ideas with some facts? Yes, we can. Giving as an acquired fact the functionality of the vertebrate blue sequence, let’s look at another interesting example: Hymenoptera.

As we can see in our table, Hymenoptera are a large order of insects which includes, in its main suborder Apocrita, three different groups of organisms: very simply, wasps, bees and ants. Now, I will not go into details of the evolutionary history of Hymenoptera, but let’s just say that the split between these three groups is reasonably old enough, let’s say in the order of 100 million years, probably more. So, we have a scenario which is similar to vertebrates: wasps, bees and ants shared a highly conserved blue sequence, and the homology has been conserved for at least about 100 million years. That should be enough to infer functionality.
However, I have tried to support the inference of significant purifying selection acting on this blue sequence, performing a Ka/Ks ratio analysis on three sequences: a wasp (Polistes canadensis), a bee (Apis mellifera) and an ant (Camponotus floridanus).
Now, this is rather technical, but I will give just the main idea. A Ka/Ks ratio is essentially the ratio of non synonymous mutations to synonymous mutations. Synonimous mutations are those which do not change the aminoacid corresponding to the codon. Therefore, they are assumed to be neutral variation. If the value of the ratio is about 1, we can usually assume that the whole sequence is under neutral variation (there are as many mutations which change the sequence as mutations which do not change it, IOWs the sequence behaves like a non functional sequence). If the value of the ratio is sgnificantly lower than 1, we usually infer negative/purifying selection: IOWs, the sequence changes less than expected if compared to neutral variation, and therefore it is reasonably under functional constraints. If the value of the ratio is higher than 1 (a rather rare case), then we usually assume positive selection on the sequence: it changes more than expected. OK, that was as simple as I could say it.
I have computed the Ka/Ks ratio for the three above mentioned sequences (blue sequences in wasp, bee and ant). Here are the results:
Wasp – Bee: Ka/Ks ratio = 0.09291813
Wasp – Ant: Ka/Ks ratio = 0.05965076
Bee – Ant: Ka/Ks ratio = 0.01145057
IOWs, the three different sequences seem definitely to be under purifying selection. Therefore, the sequences can reasonably be considered functional.
Another good example is the case of C. elegans and C. briggsae. The blue sequence in these two species, even if not too long, shows very good conservation (311 bits), and it is also equally conserved in other sequenced Caenorhabditis species (brenneri, remanei). Now, C. elegans and C. briggsae are two very similar little worms, but it is well known that they exhibit great genomic differences, and a rather old separation (maybe 100 million years again). Therefore, the conservation of the sequence in all known caenorhabditis genomes is rather telling.
So, what are the consclusions of this rather long and boring discussion? I will try to sum them up:
- In almost all Prickle 1 homologues in metazoa, a rather conserved domain sequence (the “red” part) is associated to an accessory sequence (the “blue” part) where no domain is recognizable.
- Those blue sequences are highly taxonomically restricted, and highly conserved in their restricted group of organisms, while they share almost no other homology with the rest of the existing proteome.
- There are very good reasons to infer that those sequences are functional, and that they probably contribute to the general function of the protein, and to its specificity in each group of organisms.
- So, while the red part of the molecule is a good example of function conserved through species, the blue part is a good example of function which varies completely between different groups of organisms, but is highly conserved in the gorup to which it is confined. IOWs. these blue sequences are a very good example of the concept which I have highlighted at the beginning of my previous OP:
- 2b) (Differences between homologues) can be the expression of differences in function in different species and contexts
My final and brief point is that this kind of appearance of new de novo functional sequences in different groups of organisms, their strict taxonomical restriction and their high functional constraints are all very strong arguments for a design inference. But that, of course, will be the object of the discussion which, I hope, will follow. Including mass extinctions and other lucky events. 🙂