
In the recent thread “That’s gotta hurt” Bill Cole states:
I think over the next few years 3 other origins (my note: together with OOL), will start to be recognized as equally hard to explain:
- The origin of eukaryotic cell: difficult to explain the origin of the spliceosome, the nuclear pore complex and chromosome structure.
- The origin of multicellular life: difficult to explain the origin of the ability to build complex body plans.
- The origin of man: difficult to explain the origin of language and complex thought.
That thought is perfectly correct. There are, in natural history, a few fundamental transitions which scream design more that anything else. I want to be clear: I stick to my often expressed opinion that each single new complex protein is enough to infer design. But it is equally true that some crucial points in the devlopment of life on earth certainly stand out as major engineering events. So, let’s sum up a few of them:
- OOL
- The prokaryote – eukaryote transition (IOWs, eukaryogenesis)
- The origin of metazoa (multicellular life)
- The diversification of the basic phyla and body planes (IOWs, the Cambrian explosion)
Well,  to those 4 examples, I would like to add the diversification of all major clades and subphyla.
to those 4 examples, I would like to add the diversification of all major clades and subphyla.
Of course, another fundamental transition is the one to homo sapiens, but I will not deal with it here: I fully agree with Bill Cole that it is an amazing event under all points of view, but it is also true that it presents some very specific problems, which make it a little bit different from all the other transitions we have considered above.
I will state now in advance the point that I am trying to make here: each of the transitions described requires tons and tons of new, original, highly specific functional information. Therefore, each of those transitions commands an extremely strong inference to design. I will deal in particular with the transition to the subphylum of vertebrates, for a series of reasons: being vertebrates, we are naturally specially interested in that transition; there are a lot of fully sequenced genomes and proteomes of vertebrate species ; and a lot is known about vertebrate biology. IOWs, we have a lot of data that can help us in our reasoning. So, I will try to fix a few basic points which will be the foundation of our analysis:
- a) The basic phylum is Chordates, which are characterized by the presence of a notochord. Chordates include three different clades: Craniata, Tunicata, Cephalochordata.
- b) Vertebrates are a subphylum of the phylum Chordates, and in particular of the clade Craniata. They represent the vast majority of Chordates, with about 64,000 species described. As the name suggests, they are characterized by the presence of a vertebral column, either cartilaginous or bony, which replaces the notochord.
- c) The phylum Chordate, like other phyla, can be traced at least to the Cambrian explosion (540 million years ago).
- d) Chordates which are not vertebrates are quite rare today. They include:
- 1) Craniata: the only craniates which are not vertebrates are in the class Myxini (hagfish), whose classification however remains somewhat controversial. All other craniates are vertebrates.
- 2) Tunicata (or urochordata): about 3000 species, the best known and studied is Ciona intestinalis.
- 3) Cephalochordata: about 30 species of Lancelets.
- e) The phyla most closely related to Chordates are Hemichordates (like the Acorn worm) and Echinoderms (Starfish, Sea urchins, Sea cucumbers).
- f) Vertebrates can be divided into the following two groups:
- 1) Fishes: 3 Classes:
- 1a) Jawless (lampreys)
- 1b) Cartilaginous (sharks, rays, chimaeras)
- 1c) Bony fish
- 2) Tetrapods: all the rest (frogs, snakes, birds, mammals)
- 1) Fishes: 3 Classes:
For the following analysis, I will consider vertebrates versus everything which preceded them (all metazoa, including “pre-chordates” (Hemichordates and Echinoderms) and “early chordates” (Tunicata and Cephalochordata). So, everything which is new in vertebrates had to appear in the window between early chordates and the first vertebrates: cartilaginous fish and bony fish (I will not refer to lampreys, because the data are rather scarce). So, let’s try to define the temporal window, for what it is possible:
- Chordates are already present at the Cambrian explosion, 540 my ago.
- Jawless fish appeared slightly later (about 530 my ago), but they are mostly extinct.
- The split of jawless fish into cartilaginous fish and bony fish can be traced about at 450 my ago
Therefore, with all the caution that is required, we can say that the information which can be found in both cartilaginous fish and bony fish, but not in non vertebrates (including early chordates), must have been generated in a window of less that 100 my, say between 540 my ago and 450 my ago. Now, my point is very simple: we can safely state that in that window of less than 100 million years a lot of new complex functional information was generated. Really a lot. To begin our reasoning, we can say that vertebrates are characterized by the remarkable development of two major relational systems:
- The adaptive immune system, which appears for the first time exactly in vertebrates.
- The nervous system, which is obviously well represented in all metazoa, but certainly reaches new important adaptations in vertebrates.
Mu ch can be said about the adaptive immune system, and that will probably be the object of a future OP. For the moment, however, I will discuss some aspects linked to the development of the nervous system. The only point that is important here is that the nervous system of vertebrates undergoes many important modifications, especially a process of encephalization. My interest is mainly in the developmental controls that are involved in the realization of the new body plans and structures linked to those processes. Of course, we don’t understand how those regulations are achieved. But today we know much about some molecules, especially regulatory proteins, which have an important role in the embryonal development of the vertebrate nervous system, and in particular in the development and migration of neurons, which is obviously the foundation for the achievement of the final structure and function of the nervous system. So, I will link here a recent paper which deals with some important knowledge about the process of neuron migration. I invite all those interested to read it carefully: Sticky situations: recent advances in control of cell adhesion during neuronal migration by David J. Solecki Here is the abstract:
ch can be said about the adaptive immune system, and that will probably be the object of a future OP. For the moment, however, I will discuss some aspects linked to the development of the nervous system. The only point that is important here is that the nervous system of vertebrates undergoes many important modifications, especially a process of encephalization. My interest is mainly in the developmental controls that are involved in the realization of the new body plans and structures linked to those processes. Of course, we don’t understand how those regulations are achieved. But today we know much about some molecules, especially regulatory proteins, which have an important role in the embryonal development of the vertebrate nervous system, and in particular in the development and migration of neurons, which is obviously the foundation for the achievement of the final structure and function of the nervous system. So, I will link here a recent paper which deals with some important knowledge about the process of neuron migration. I invite all those interested to read it carefully: Sticky situations: recent advances in control of cell adhesion during neuronal migration by David J. Solecki Here is the abstract:
The migration of neurons along glial fibers from a germinal zone (GZ) to their final laminar positions is essential for morphogenesis of the developing brain, aberrations in this process are linked to profound neurodevelopmental and cognitive disorders. During this critical morphogenic movement, neurons must navigate complex migration paths, propelling their cell bodies through the dense cellular environment of the developing nervous system to their final destinations. It is not understood how neurons can successfully migrate along their glial guides through the myriad processes and cell bodies of neighboring neurons. Although much progress has been made in understanding the substrates (1–4), guidance mechanisms (5–7), cytoskeletal elements (8–10), and post-translational modifications (11–13) required for neuronal migration, we have yet to elucidate how neurons regulate their cellular interactions and adhesive specificity to follow the appropriate migratory pathways. Here I will examine recent developments in our understanding of the mechanisms controlling neuronal cell adhesion and how these mechanisms interact with crucial neurodevelopmental events, such as GZ exit, migration pathway selection, multipolar-to-radial transition, and final lamination.
In brief, the author reviews what is known about the process of neuronal cell adhesion and migration. Starting from that paper and some other material, I have chosen a group of six regulatory proteins which seem to have an important role in the above process. They are rather long and complex proteins, particularly good for an information analysis. Here is the list. I give first the name of the protein, and then the length and accession number in Uniprot for the human protein:
- Astrotactin 1, 1302 AAs, O14525
- Astrotactin 2, 1339 AAs, O75129
- BRNP1 (BMP/retinoic acid-inducible neural-specific protein 1), 761 AAs, O60477
- Cadherin 2 (CADH2), 906 AAs, P19022
- Integrin alpha-V, 1048 AAs, P06756
- Neural cell adhesion molecule 1 (NCAM1), 858 AAs, P13591
This is a very interesting bunch of molecules:
- Astrotactin 1 and 2 are two partially related perforin-like proteins. ASTN-1 is a membrane protein which is directly responsible for the formation of neuron–glial fibre contacts. ASTN2 is not a neuron-glial adhesion molecule, but it functions in cerebellar granule neuron (CGN)-glial junction formation by forming a complex with ASTN1 to regulate ASTN1 cell surface recruitment. More about these very interesting proteins can be found in the following paper:
Structure of astrotactin-2: a conserved vertebrate-specific and perforin-like membrane protein involved in neuronal development by Tao Ni, Karl Harlos, and Robert Gilbert
- BRNP1 is another protein which functions in neural cell migration and guidance
- Cadherin 2, or N-cadherin, is active in many neuronal funtions and in other tissues, and seems to have a crucial role in glial-guided migration of neurons
- Integrin alpha-V, or Vitronectin receptor, is one of the 18 alpha subunits of integrins in mammals. Integrins are transmembrane receptors that are the bridges for cell-cell and cell-extracellular matrix (ECM) interactions.
- NCAM1 is a cell adhesion molecule involved in neuron-neuron adhesion, neurite fasciculation, outgrowth of neurites
Now, why have I chosen these six proteins, and what do they have in common? They have two important things in common:
- They are all big regulatory proteins, and they are all involved in a similar regulatory network which controls endocytosis, cell adhesion and cell migration in neurons, and therefore is in part responsible for the correct development of the vertebrate nervous system
- All those six proteins present a very big informarion jump between pre-vertebrate organisms and the first vertebrates
The evolutionary history of those six protein is summarized in the following graph, realized as usual by computing the best homology bit score with the human protein in different groups of organisms.
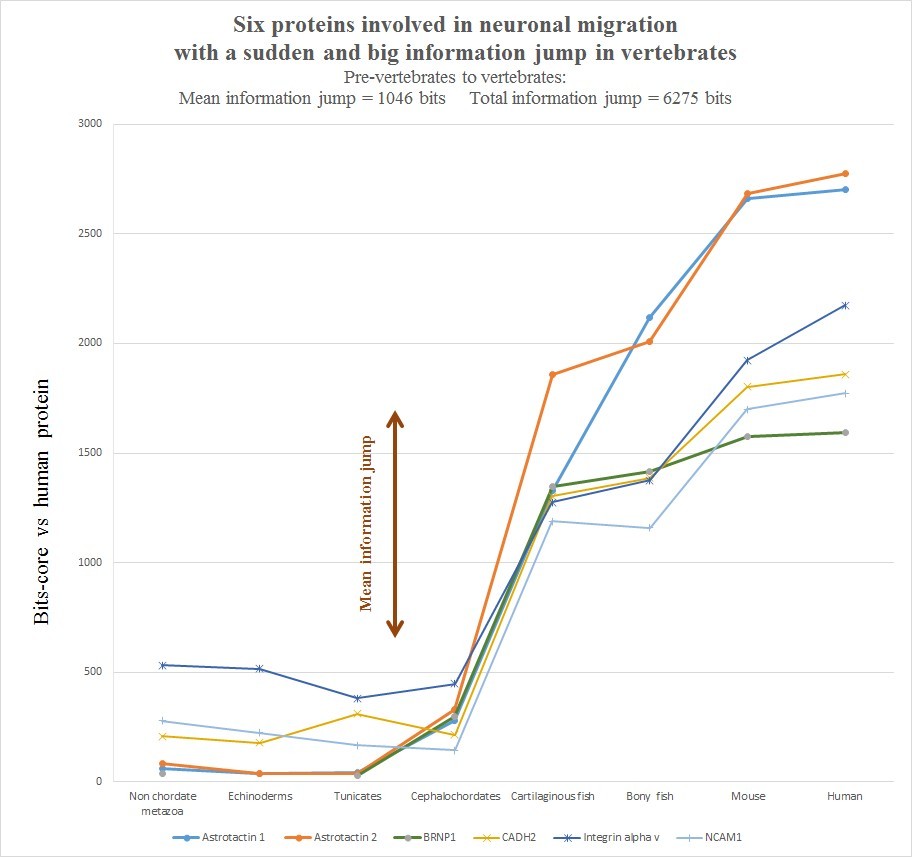
Very briefly, all the six human molecules have low homology with pre-vertebrates, while they already show a very high homology in cartilaginous fishes. The most striking example is probably Astrotactin 2, which presents the biggest jump from cephalochordata (329 bits) to cartilaginous fishes (1860 bits), for a great total of 1531 bits of jump! The range of individual jumps in the group is 745 – 1531 bits, with a mean jump of 1046 bits per molecule and a total jump of 6275 bits for all six molecules. The jump has always been computed as the difference between the best bit score in cartilaginous fishes and the best bitscore in all pre-vertebrate metazoa. We can also observe that the first three proteins have really low homology with everything up to tunicates, but show a definite increase in Cephalochordata, which precedes the big jump in cartilaginous fishes, while the other three molecules have a rather constant behaviour in all pre-vertebrate metazoa, with a few hundred bits of homology, before “jumping” up in sharks. One could ask: is that a common behaviour of all proteins? The answer is no. Look at the following graph, which shows the same evolutionary history for two other proteins, both of them very big regulatory proteins, both of them implied in the same processes as the previous six.
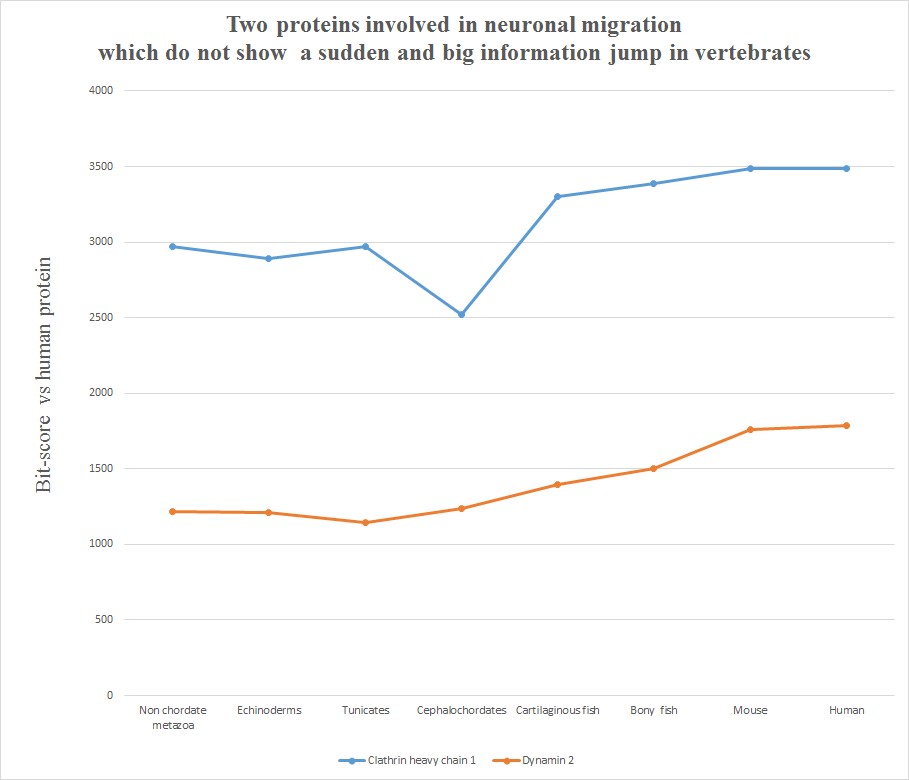
Here, the behaviour is completely different. While there is a slight increase of homology in time, with a few smaller “jumps”, there is nothing comparable to the thousand bit jumps in the first six molecules. IOWs, these two molecules already show a very high level of homology to the human form in pre-vertebrates, and change only relatively little in vertebrates. We can say, therefore, that most of the functional information in these two proteins was already present before the transition to vertebrates.
So, to sum up:
- a) The six proteins analyzed here all exhibit a huge informational jump between pre-vertebrates and vertebrates. The total functional informational novelty for just this small group of proteins is more than 6000 bits, with a mean of more than 1000 bits per protein.
- b) These proteins are probably crucial agents in a much more complex regulation network implied in neuron adhesion, endocytosis, migration, and in the end in the vast developmental process which makes individual neurons migrate to their specific individual locations in the vertebrate body plan.
- c) The above process is certainly much more complex than the six proteins we have considered, and implies other proteins and obviously many non coding elements. Our six proteins, therefore, can be considered as a tiny sample of the general complexity of the process, and of the informational novelty implied in the process itself.
- d) Moreover, the process regulating neuron migration is certainly strictly integrated, with so many agents working in a coordinated way. Therefore, there is obviously a strong element of irreducible complexity implied in the whole informational novelty of the vertebrate process, an element that we can only barely envisage, because we still understand too little.
- e) The neuron regulation process, of course, is only a part of the informational novelty implied in vertebrates, a small sample of a much more complex reality. For example, there is a lot of similar novelty implied in the workings of the immune system, of the cytokine signaling system, and so on.
- f) The jump described here is really a jump: there is no trace of intermediate forms which can explain that jump in all existing pre-vertebrates. Of course, neo darwinists can always dream of lost intermediates in extinct species. This is a free world.
- g) Are these 6000+ bits of functional information really functional? Yes, they are. Why? because they have been conserved for more than 400 million years. Remember, the transition we have considered happens between the first chordates and cartilaginous fish, and it can be traced to that range of time. And those 6000+ bits are bits of homology between cartilaginous fish and humans.
- h) How much is 6000 bits of functional information? It is really a lot! Remember, Dembski’s Universal Probability Bound, taking in consideration the whole reasonable probabilistic resource of our whole universe from the Big Bang to now, is just 500 bits. 6000 bits correspond to a search space of 2^6000, IOWs about 10^2000, a number so big that we cannot even begin to visualize it. It’s good to remind ourselves, from time to time, that we are dealing with exponential values.
- i) How great is the probability that 6000 bits of functional information can be generated in a window time of less than 100 million years, by some unguided process of RV + NS in six objects connected in an irreducibly complex system, even if RV were really helped by some NS in intermediates of which there is no trace? The answer is simple: practically non existent.
- j) Therefore, the tiny sample of six proteins that we have considered here, which is only a small part of a much bigger scenario, points with extreme strength to a definite design inference:
The transition to vertebrates was a highly engineered process. The necessary functional information was added by design.