 |
Could proteins have developed naturally on Earth, without any intelligent guidance? The late astrophysicist Sir Fred Hoyle (1915-2001) thought not, and one can immediately grasp why, just by looking at the picture above, which shows the protein hexokinase, with much smaller molecules of ATP and the simplest sugar, glucose, shown in the top right corner for comparison (image courtesy of Tim Vickers and Wikipedia). Briefly, Hoyle argued that since a protein is typically made up of at least 100 or so amino acids, of which there are 20 kinds, the number of possible amino acid sequences of length 100 is astronomically large. Among these, the proportion that are able to fold up and perform a biologically useful task as proteins is vanishingly small. Hoyle argued that there wouldn’t have been enough time for Nature to explore the set of all possibilities on the primordial Earth and hit on a protein that could do something useful. Even billions of years would not have been enough. The origin of even a single protein looks like a biochemical miracle. I should mention in passing that there are a lot of misleading Web sites on “Hoyle’s fallacy”, which purport to take apart his argument without a proper understanding of the mathematical logic that underlies it. For those who would like to learn more about Hoyle’s argument, I would recommend biologist Stephen Jones’ online article, Fred Hoyle about the 747, the tornado and the junkyard, as well as a 1981 essay by Hoyle entitled, The Universe: Past and Present Reflections. But I digress.
Over at the Skeptical Zone, Allan Miller has written a post, entitled, Journal club – Protein Space. Big, isn’t it?, which attempts to refute the claim made by the late Sir Fred Hoyle (1915-2001) that the chance of Nature hitting on a functional protein during the 4.5-billion-year history of the Earth is astronomically low. Miller argues that there would have been plenty of time for Nature to build functional proteins on the primordial Earth. Evolution, he claims, could still work perfectly well even if the set of all “possible proteins” were much smaller than it is today, making it easier to explore quickly; moreover, evolution could have easily searched this reduced set of all “possible proteins” within the time available (say, a billion years), and hit upon some proteins that actually worked. In his post, Miller also addresses the thermodynamic issues relating to how proteins could have formed in the first place.
To support his case, Miller cites a 2008 paper by David Dryden, Andrew Thomson and John White of Edinburgh University, entitled, How much of protein sequence space has been explored by life on Earth?, which defends the claim that “a reduced alphabet of amino acids is quite capable of producing all protein folds (approx. a few thousand discrete folds; Denton 2008) and providing a scaffold capable of supporting all protein functions…. Therefore it is entirely feasible that for all practical (i.e. functional and structural) purposes, protein sequence space has been fully explored during the course of evolution of life on Earth.”
Miller may or may not be aware that there have been no less than four responses to Dryden, Thomson and White’s paper, which shoot it full of holes.
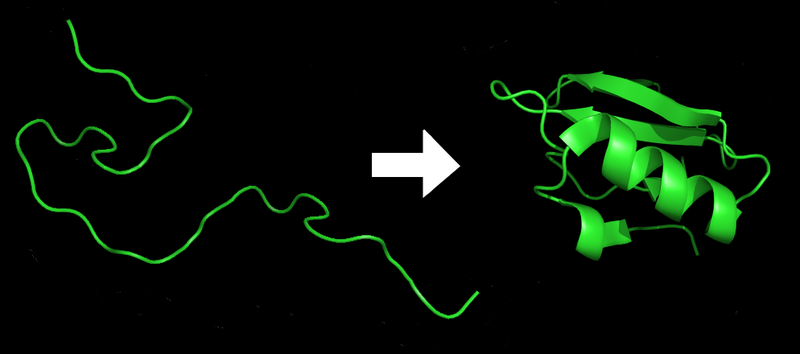 |
Proteins before and after folding. Image courtesy of Wikipedia.
1. A response to Dryden, Thomson and White from Dr. Cornelius Hunter
Dr. Cornelius G. Hunter is a graduate of the University of Illinois where he earned a Ph.D. in Biophysics and Computational Biology. In 2011, Dr. Hunter rebutted the arguments in Dryden, Thomson and White’s paper, in two posts over at his blog, Darwin’s God: Response to Comments: Natural Selection Doesn’t Help, Gradualism is Out, and so is Evolution (July 2, 2011) and The Amyloid Threat, Big Numbers Game and Quote Mining: Protein Evolution and How Evolutionists Respond to the Empirical Evidence (September 15, 2011). The key points from Dr. Hunter’s response are as follows:
The paper [by Dryden, Thomson and White] attempts to make two general points. First that evolution can succeed with a much smaller protein sequence space and second, that evolution can easily search the entire protein sequence space. Both conclusions are scientifically ridiculous and are inconsistent with what we do understand about proteins…
For the first claim, the evolutionists argue for a smaller protein sequence space because:
A. “the actual identity of most of the amino acids in a protein is irrelevant” and so we can assume there were only a few amino acids in the evolution of proteins, rather than today’s 20.B. Only the surface residues of a protein are important.
C. Proteins need not be very long. Instead of hundreds of residues, evolution could have used about 50 for most proteins.
For Point A, the evolutionists use as support a series of simplistic studies that replaced the actual protein three-dimensional structure and amino acid chemistries with cartoon, two-dimensional lattice versions.…
Likewise Point B is at odds with science, and again is an unwarranted extrapolation on a simplistic lattice study.
For Point C, the evolutionists note that many proteins are modular and consist of self-contained domains “of as few as approximately 50 amino acids.” But the vast majority of protein domains are far longer than 50 residues. Single domain proteins, and domains in multiple-domain proteins are typically in the hundreds of residues…
To defend their second claim, that evolution can easily search the entire protein sequence space, the evolutionists present upper and lower bound estimates of the number of different sequences evolution can explore.
Their upper bound estimate of 10^43 (a one followed by 43 zeros) is ridiculous. It assumes a four billion year time frame with 10^30 bacteria constantly testing out new proteins… You can’t use bacteria to explain how proteins first evolved when the bacteria themselves require an army of proteins.
The lower bound of 10^21 is hardly any more realistic. The evolutionists … continue to rely on the pre-existence of an earth filled with a billion species of bacteria (with their many thousands of pre-existing proteins)…
The scientific fact is that the numbers are big. This isn’t a “game.”
For instance, consider an example protein of 300 residues (many proteins are much longer than this). With 20 different amino acids to choose from, there are a total of 10^390 different amino acid sequences possible. Now let’s simplify and assume only four different amino acids are needed. This reduces the problem to 10^180 different sequences.
Next let’s assume that only 50% of the residues are important. At the other 50%, any amino acid will do. That is, fully half of the amino acid sequence is inconsequential. These are extremely aggressive and unrealistic assumptions, yet nonetheless we are left with a total of 10^90 sequences. 90 may not appear to be a big number, but a one followed by 90 zeros is. It is completely impractical for evolution.
And if you don’t agree with my example, then we have the evolutionary experiments, described above, which concluded that 10^70 tries would be required. And even that was only for a fraction of the protein machine, and it assumed a pre-existing biological world with its many proteins already in place.
So let’s take the evolutionist’s own numbers at face value, giving them every advantage. The number of experiments required is 10^70 and the number of experiment possible is 10^43. Even here, giving the evolutionists every advantage, evolution falls short by 27 orders of magnitude.
The theory, even by the evolutionist’s own reckoning, is unworkable. Gradualistic evolution—the test that Darwin himself set forth—or non gradualistic evolution, it does not matter. Evolution fails by a degree that is incomparable in science.
The numbers, then, appear to rule out the scenario envisaged by Dryden, Thomson and White. Even using the wildly optimistic suppositions made by Darwinian evolutionists, there wouldn’t have been enough time for Nature to try out all possibilities and thereby hit upon a sequence of amino acids that could fold up properly, enabling it to perform a biologically useful task. Billions of years isn’t anywhere near enough time, when you need decillions of years to complete the task!
2. A response to Dryden, Thomson and White from Dr. Douglas Axe
Miller also appears to be unaware that Biologic Institute director Dr. Douglas Axe responded to Dryden, Thomson and White’s paper back in 2008, in an online post entitled, Science stories. Dr. Axe’s credentials in the field are impressive: after obtaining a Caltech Ph.D., he held postdoctoral and research scientist positions at the University of Cambridge, the Cambridge Medical Research Council Centre, and the Babraham Institute in Cambridge. He has also written two articles for the Journal of Molecular Biology (see here and here for abstracts). He has also co-authored an article published in the Proceedings of the National Academy of Sciences, an article in Biochemistry and an article published in PLoS ONE. His work has been reviewed in Nature and featured in a number of books, magazines and newspaper articles.
Dr. Axe’s article pithily summarizes of Dryden, Thomson and White’s argument before proceeding to mow it down:
Here we’ll look at a recent paper by Dryden, Thomson, and White (DTW), published in Journal of the Royal Society Interface. [2] Its stated conclusion is: “It is entirely feasible that for all practical (i.e. functional and structural) purposes, protein sequence space has been fully explored during the course of evolution of life on Earth.” What this means, in simple language, is that the functions we see proteins performing in cells are not so extraordinary that we should be surprised to see them.
To understand how the DTW [Dryden, Thomson and White – VJT] paper attempts to justify its claim, consider the following analogy between proteins and sentences. Just as sentences are written by arranging characters in sequence, so proteins are built by linking amino acids into strings with specified sequences. The amino acid ‘alphabet’ has twenty members, comparable to the size of actual alphabets, and the length of protein ‘sentences’ written in their alphabet is similar to the length of actual written sentences. In both cases the ability to do many useful things by arranging characters into appropriate sequences opens up a world of possibilities.
By this way of viewing things, cells depend on several thousand protein ‘sentences’, each with its own important meaning. Considering the complexity of this biological ‘text’, chance-based explanations of it certainly call for careful probabilistic evaluation. But as DTW point out, the actual probabilistic difficulty of such a thing depends on several factors. Their paper focuses on two of these: the length of the required ‘sentences’, and the size of the ‘alphabet’ needed to write them. Their claim is that neither of these requirements is really as stringent as it appears to be.
We’ll use the analogy to get a feel for this claim, keeping in mind of course that the claim is about proteins rather than sentences. Consider the DTW conclusion quoted above. That sentence is 185 characters long, making it similar in length to biological proteins [3]…
We might try shortening it to “Earthly life has fully explored protein functions.” This brings the length down to 50 characters, though not without affecting the meaning. The bigger problem, though, is that the DTW proposal also calls for radical reduction of the alphabet size. In fact, for this shortened sentence to meet their proposal, we would need to re-write it with a tiny alphabet of four or five symbols — and that mini-alphabet would have to work not just for this sentence but for all sentences in a text the size of the DTW paper.
…According to DTW, the functions that biological proteins perform could be adequately performed with proteins that are much shorter and incorporate considerably fewer kinds of amino acids…
In fact, the most conclusive scientific evidence on this matter seems to contradict their claim. First and foremost is the very observation they seek to explain — the functional proteins we see in nature. The mere fact that these proteins are far too long and employ far too many amino acids to meet the DTW restrictions ought to make us assume that they don’t meet those restrictions, absent a convincing case that they do. After all, why would cells go to so much trouble making all twenty amino acids if far fewer would do? And if fewer really would do, why do cells so meticulously avoid mistaking any of the twenty for any other in their manufacture of proteins? [4]
The cellular apparatus for making proteins does incorporate wrong amino acids, but the rarity of these errors makes the process remarkably well tuned for accurate synthesis of long proteins, not mini-proteins. A popular biochemistry textbook puts it this way: “An error frequency of about 0.0001 per amino acid residue was selected in the course of evolution to accurately produce proteins consisting of as many as 1000 amino acids while maintaining a remarkably rapid rate for protein synthesis.” [5] So, while the textbook ignores the problem that the DTW paper addresses — how on earth such things could evolve — the DTW paper ignores the aspects of proteins that plainly defy simplification.
What’s more, the inherent complexity of biological proteins is confirmed by experiments that test it directly. We know that amino acid changes tend to be functionally disruptive even when the replacements are similar to the originals [6], and we know what typically causes this — reduced structural stability of the functional form [7]. So, not only does the DTW claim suffer from a lack of direct supportive evidence — it also suffers from a substantial body of directly contrary evidence.
References
[2] http://rsif.royalsocietypublishing.org/content/5/25/953.full
[3] http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1150220
[4] http://www.sciencedirect.com/science
[5] Berg JM, Tymoczko JL, Stryer L (2002) Biochemistry (5th edition). Freeman.
Dr. Axe’s questions deserve to be answered: why would Nature go to the trouble of building proteins out of 20 different kinds of amino acids, if just a few would suffice? And why would it come up with an efficient mechanism for detecting errors in amino acids, if these errors don’t matter very much?
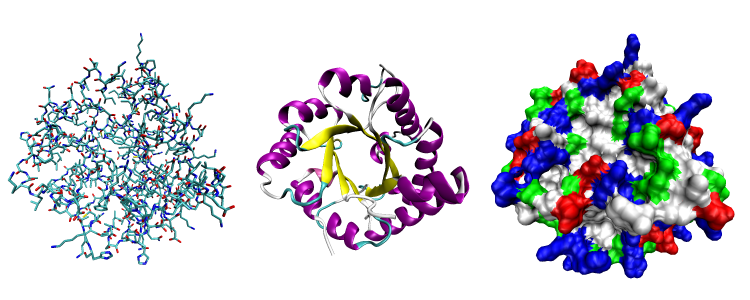 |
Three possible representations of the three-dimensional structure of the protein triose phosphate isomerase. Left: all-atom representation colored by atom type. Middle: Simplified representation illustrating the backbone conformation, colored by secondary structure. Right: Solvent-accessible surface representation colored by residue type (acidic residues red, basic residues blue, polar residues green, nonpolar residues white). Image courtesy of Wikipedia.
3. Dr. Kirk Durston’s criticisms of Dryden, Thomson and White’s paper
In 2009, another critic, Dr. Kirk Durston, uncovered several serious flaws in Dryden, Thomson and White’s paper. Dr. Kirk Durston completed his Ph.D. in Biophysics at the University of Guelph, specializing in the identification, quantification, and application of functional information to protein structure. He is also the Director of the New Scholars Society. Dr. Durston sent a letter to Dryden, who promised that he would get back to Durston, after initially corresponding with him. Unfortunately, he never did. Dr. Durston has kindly forwarded me a copy of his original letter of March 2, 2009, from which I quote the following excerpts:
Sir,
I read your brief paper with interest. You have presented some interesting ideas, but I do have some major reservations which I have summarized below.
1. If one reduces the size of sequence space by reducing its dimensionality, the size of functional sequence space is also reduced. Of course, your point was that if the size of sequence space was sufficiently reduced, then all of the remaining sequence space could be searched given certain parameters. My concern has to do with whether functional sequence space shrinks to zero before the size of the total sequence space becomes small enough to be adequately explored. My own research suggests that it might be radically optimistic to posit that 2-amino acid space (or 2-property space), or even 3-amino acid space contains any functional sequence space for the majority of protein families…
2. It is true that if one examines a sequence alignment for an entire protein family, often consisting of 1,000 or more sequences, no site is perfectly conserved. That observation can be misleading, however, for it ignores the higher order associations between sites. These higher order relationships, related to the final structure and function of the protein, often require that if amino acid x occurs at site b, then amino acid y is required at site g and amino acid z is required at site k… The bottom line is this: functional sequence space is much smaller than what one might infer from simply looking at the variation in amino acids per site, due to the higher order associations between sites.…
3. My current research involves the computational detection of higher order relationships between sites… [T]he protein family that I have been analyzing contains numerous higher order site relationships ranging from simple pair-wise relationships up to one 29th order relationship (consists of 29 sites). In mapping these higher order relationships to the 3-D structure of that protein, it can be seen that these relationships are structural. My point is that any discussion of functional sequence space must take into consideration the higher order relationships within the protein that are essential to the stability of its structure. Given what I am seeing in my own research, the size of functional sequence space for most proteins is likely to shrink to zero before the overall sequence space is downsized to 2 or even 3-amino acid space (or 2-property space) is reached. For this reason, I do not think a hypothesized 2 or 3-amino acid space is going to be sufficient for most of the protein families.
4. With regard to whether protein families can be simplified to one polar and one non-polar amino acid, or even two groups of acids, one polar and one non-polar, again my results suggest otherwise. I have attached an Excel file that contains my results for Ribosomal S2, a universal protein and, thus, possibly a component of the LUCA [last universal common ancestor – VJT]… These results suggest that a simple polar/non-polar 2-amino acid world would be a severe problem for RS 2. I have similar findings for RS 7, another universal protein. In the early stages of my research, my hypothesis was that the universal proteins were likely to be less complex and easier to find in an evolutionary search. I began with transforming their 20-amino acid sequence space into 2-property sequence space (as you have proposed). It became apparent in very short order that 2- property space was far too crude.…
5. I would think that a prediction arising out of your hypothesis would be that the universal proteins should be more amenable to a 2 or a 3-amino acid world, (or a 2-property world) if we assume they are required for LUCA and are, thus, quite early. I’ve looked at several universal proteins and I am not optimistic that this prediction can survive falsification… Alternatively, your hypothesis predicts a relatively smooth probability distribution for the 20 amino acids. I’ve computed the probability distributions for all 20 amino acids in 35 different protein families. They are not even close to a 2-property probability distribution.
6. Finally, I see massive problems in going from, say, a 3-amino acid biological world to a 20 amino acid world. For example, as I’m sure you readily recognize, there are large problems in going from coding for 3 amino acids to coding for 20 amino acids. I am sure you are aware of several other very obvious problems related to the coding, translation, and fitness process. As you know, many of the universal proteins are related to the ribosome/translation component of biological life. From a purely Darwinian perspective, there is an energy and fitness expense for having 20 amino acids if just 2 or 3 or 5 will do.…
Overall, my work with real data involving 35 different protein families, has left me not at all optimistic that the hypothesis you advance in your paper is viable… What I am seeing is that the functional complexity of the coding, structure and function of protein families is significantly more advanced than I previously expected.
Best regards,
Kirk
Computational Biophysics, University of Guelph
It seems that Dryden, Thomson and White’s paper has died the death of a thousand cuts at the hands of Dr. Durston’s skilled, rapier-like logic. Proteins are complex beasties, whose parts are delicately inter-linked. The idea that they could retain their functionality – or indeed, any kind of functionality – while the number of different kinds of amino acids they contain is reduced from 20 to 2 or 3 is simply preposterous, as well as being at odds with the experimental data that’s available to date. Moreover, there would have been an enormous cost in Nature’s using 20 amino acids to build a functional protein, if just a few would do the trick.
I’d like to mention one more critic of Dryden, Thomson and White’s paper, who has administered what I consider to be the coup do grace with his work on “singleton” proteins. “Singleton proteins?” I hear you ask. “What are they?” Suffice to say that you’ll be hearing a lot more about them from now on, on this Website. Let us continue.
4. Dr. Branko Kozulic’s rebuttal of Dryden, Thomson and White’s paper
Dr. Branko Kozulic received Ph.D. in biochemistry from the University of Zagreb, Croatia, in 1979. From 1983 to 1988 he worked at Institute of Biotechnology, ETH-Zurich, Switzerland. For about fifteen years he was employed at a private Swiss biotech company, of which he was a co-founder. He currently works at Gentius Ltd. in Zadar, Croatia. He also teaches at the Faculty of Food Technology and Biotechnology in Zagreb. He is also a member of the Editorial Team of the journal Bio-complexity.
In 2011, Dr. Kozulic authored a paper entitled, Proteins and Genes, Singletons and Species. In his paper, Dr. Kozulic discusses the difficulty of generating even one functional protein by a random search, during the Earth’s 4.5 billion-year history. He then proceeds to assess the claims made in Dryden, Thomson and White’s paper, which argues that the search for the Earth’s first proteins may have been far easier than it looks today, as proteins were shorter then and were composed of fewer amino acids:
One strategy for defusing the problem associated with the finding of functional proteins by random search through the enormous protein sequence space has been to arbitrarily reduce the size of that space. Because the space size is related to protein length (L) as 20^L, where 20 denotes the number of different amino acids of which proteins are made, the number of unique protein sequences will rapidly decrease if one assumes that the number of different amino acids can be less than 20. The same is true if one takes small L values. Dryden et al. used this strategy to illustrate the feasibility of searching through the whole protein sequence space on Earth, estimating that the maximal number of different proteins that could have been formed on planet Earth in geological time was 4 x 10^43 [9]. In [the] laboratory, researchers have designed functional proteins with fewer than 20 amino acids [10, 11], but in nature all living organisms studied thus far, from bacteria to man, use all 20 amino acids to build their proteins. Therefore, the conclusions based on the calculations that rely on fewer than 20 amino acids are irrelevant in biology. Concerning protein length, the reported median lengths of bacterial and eukaryotic proteins are 267 and 361 amino acids, respectively [12]. Furthermore, about 30% of proteins in eukaryotes have more than 500 amino acids, while about 7% of them have more than 1,000 amino acids [13]. The largest known protein, titin, is built of more than 30,000 amino acids [14]. Only such experimentally found values for L are meaningful for calculating the real size of the protein sequence space, which thus corresponds to a median figure of 10^347 (20^267) for bacterial, and 10^470 (20^361) for eukaryotic proteins.
Kozulic’s take-home message is clear and unambiguous: in the real world, proteins need 20 different amino acids, not two. Moreover, hundreds of amino acid molecules are required to make a typical protein.
Now, this wouldn’t be so bad if proteins were somehow bunched or linked together in terms of their chemical properties. That way, you might start with a small functional protein and (through a series of lucky chemical accidents) eventually work your way up from that small protein to a larger one, via a kind of “island-hopping” process. But proteins in the real world aren’t like that. Many of them are loners, with no close chemical “relatives.” Such proteins are called “singletons”, and they make up a very large proportion of all known proteins.
What have we learned from these tens of millions of protein sequences originating from the genomes of more than one thousand species? When proteins of similar sequences are grouped into families, their distribution follows a power-law [65-72], prompting some authors to suggest that the protein sequence space can be viewed as a network similar to the World Wide Web, electrical power grid or collaboration network of movie actors, due to the similarity of respective distribution graphs. There are thus small numbers of families with thousands of member proteins having similar sequences, while, at the other extreme, there are thousands of families with just a few members. The most numerous are “families” with only one member; these lone proteins are usually called singletons. This regularity was evident already from the analysis of 20 genomes in 2001 [66], and 83 genomes in 2003 [69]. As more sequences were added to the databases more novel families were discovered, so that according to one estimate about 180,000 families were needed for complete coverage of the sequences in the Pfam database from 2008 [71]. Another study, published in the same year, identified 190,000 protein families with more than 5 members – and additionally about 600,000 singletons – in a set of 1.9 million distinct protein sequences [73]
References
65. Huynen MA, van Nimwegen E (1998) The frequency distribution of gene family sizes in complete genomes. Mol Biol Evol 15: 583-589.
66. Qian J, Luscombe NM, Gerstein M (2001) Protein family and fold occurrence in genomes: power-law behaviour and evolutionary model. J Mol Biol 313: 673-681. doi:10.1006/jmbi.2001.5079.
67. Luscombe NM, Qian J, Zhang Z, Johnson T, Gerstein M (2002) The dominance of the population by a selected few: power-law behaviour applies to a wide variety of genomic properties. Genome Biol 3:research0040.1-0040.7.
68. Unger R, Uliel S, Havlin S (2003) Scaling law in sizes of protein sequence families: from super-families to orphan genes. Proteins 51: 569-576.
69. Enright AJ, Kunin V, Ouzounis CA (2003) Protein families and TRIBES in genome sequence space. Nucleic Acids Res 31: 4632-4638. doi:10.1093/nar/gkg495.
70. Lee D, Grant A, Marsden RL, Orengo C (2005) Identification and distribution of protein families in 120 completed genomes using Gene3D. Proteins 59: 603-615. doi:10.1002/prot.200409.
71. Sammut SJ, Finn RD, Bateman A (2008) Pfam 10 years on: 10 000 families and still growing. Brief Bioinform 9: 210-219. doi:10.1093/bib/bbn010.
72. Orengo CA, Thornton JM (2005) Protein families and their evolution – a structural perspective. Annu Rev Biochem 74: 867-900. doi:10.1146/annurev.biochem.74.082803.133029.
73. Yeats C, Lees J, Reid A, Kellam P, Martin N, Liu X, Orengo C (2008) Gene3D: comprehensive structural and functional annotation of genomes. Nuclei Acids Res 36: D414-D418. doi: 10.1093/nar/gkm1019.
The take-home message here is that the island-hopping strategy won’t work: most of the proteins that exist in Nature are “loners” or “singletons”, that can’t be generated in this way. Intelligent foresight is the only known process that can overcome this probabilistic hurdle. The evidence from what we know about proteins is by now luminously clear: they could only have been made by careful planning.
Thermodynamic issues relating to protein formation
When discussing thermodynamic issues relating to the formation of the first proteins on the primordial Earth, Allan Miller makes some rather remarkable concessions in his post over at The Skeptical Zone. He frankly acknowledges that proteins could never have evolved in the Earth’s primordial seas (Darwin’s “warm little pond”):
The ‘warm little pond’ is chemically naive; a strawman. Darwin (who coined the phrase) knew nothing of thermodynamics, nor protein. The free energy change associated with condensation/hydrolysis of the peptide bond means that it requires the input of energy to make it. The energy of motions of molecules in solution is not enough. Even with appropriate energy, having hit the jackpot once is insufficient. One has to retain that sequence, and this random process is not repeatable. So calculating ‘the probability of a protein’ by combinatorial means is irrelevant if that is not how it happened.
Allan Miller proposes instead that ribozymes may have played a role in assisting peptide chains to explore the whole of protein space.
Like short peptides, short RNA and DNA strands have catalytic ability (ribozymes), and all the basic reactions are within their scope. One particularly relevant reaction is the ability to join an amino acid to a nucleic acid monomer, ATP, which can be accomplished by a ribozyme just 5 bases long. This is a central step in modern protein synthesis, the lone monomer now extended by an elaborate ‘tail’ arrangement – the tRNA molecule – and the joining now performed by a protein catalyst. Charging the acid in this way overcomes the thermodynamic barrier to peptide synthesis, because aminoacylated ATP has the energy to form a peptide bond where ‘bare’ amino acids do not. This gives an inkling of the mode by which Hoyle’s peptide space may have been actually accessed and explored. Short peptides formed by ribozymes from a limited acid library, with limited catalytic ability, may become longer and more specific and versatile by duplications and recombination of subunits.
I’d like to answer Miller’s proposal with a single picture. This is what a ribozyme looks like. It’s the hammerhead ribozyme (image courtesy of William G. Scott and Wikipedia).
I put it to my readers that Miller has solved one problem (how to build proteins) only by creating another (how to build ribozymes).
I’d also like to quote what the Wikipedia article says about this ribozyme:
In the natural state, a hammerhead RNA motif is a single strand of RNA, and although the cleavage takes place in the absence of enzymes, the hammerhead RNA itself is not a catalyst in its natural state, as it is consumed by the reaction and cannot catalyze multiple turnovers.
Hmmm. Doesn’t sound too promising, does it? And on top of that, it appears fiendishly difficult to generate naturally, in the absence of intelligent guidance.
But don’t take my word for it. I suggest that readers go and have a look at the article, A New Study Questions RNA World (April 16, 2012) over at Evolution News and Views:
A new study in PLoS One shows that RNA and the proteins involved in protein synthesis must have co-evolved. This flies in the face of RNA-world theories, which presume that RNA formed first and that catalytic function (usually performed by proteins) was completed by catalytic RNA, known as ribozymes.
Researchers at the University of Illinois used phylogenetic modeling methods to evaluate the evolutionary history of the ribosome by correlating RNA structure and the ribosome protein structure. Their studies reveal several things of interest.
One of the assumptions in the RNA first hypothesis is that the active site of the ribosome, the peptidyl transferase center (PTC), which is the key player in protein synthesis, evolved first. However, Harish et al.’s studies reveal that the ribosome subunits actually evolved before the PTC active site and those subunits co-evolved with RNA, or what would eventually be sections of tRNA.
The authors conclude that their study answers some of the difficult questions associated with the RNA First World, while suggesting that there may have been a ribonuceloprotein primordial world…
Assessment
Overall, the authors appeal to co-option and co-evolution and justify this using phylogenetic homology studies. They contend as many in the ID camp do that “the de novo appearance of complex functions is highly unlikely. Similarly, it is highly unlikely that a multi-component molecular complex harboring several functional processes needed for modern translation could emerge in a single or only a few events of evolutionary novelty.” Their explanation, however, is that a simpler system was performing a different function, and then was recruited into the complex protein translation machine.
The question that follows is what exactly did the recruiting? What provokes recruitment to another system? The authors labeled this time of recruitment the “first major transition” but their explanation of the transition is a little cloudy.
They seem to answer the question of “motivation to recruitment” by appealing to co-evolution. The RNA and ribosome proteins are co-dependent such that as one evolves, the other does too and somehow it reached a point where a “major transition” occurs.
There are many striking features of this study, such as the authors’ acknowledgement of the deficiency of ribozymes to account for the “chicken-and-egg” problem with protein synthesis, and their recognition of the improbable evolution of RNA apart from the ribosomal protein in view of the fact that the relevant functions are so intimately intertwined.
While these results show a relationship and even a correlation between tRNA and the ribosome, it is still unclear what exactly promoted recruitment, what attracted the tRNA to the proto-ribosome, or why co-option must be the conclusion. Could this not also be a case of an irreducibly complex machine?
Indeed. In the absence of any experimental data confirming these fanciful speculations about how proteins may have arisen via an unguided natural process, I can only regard them as the chemical equivalent of castles in the air.
But speculation is one thing; misinformation is another. Even though there are now several detailed online rebuttals of claims Dryden, Thomson and White’s claim that there would have been plenty of time for natural processes to hit upon a functional protein on the primordial Earth, the myth refuses to die. It is to be hoped that this little bouquet of mine, in which I have brought all the rebuttals together in a single post, will help to slay this myth.
All the evidence we have to date on proteins points towards a single conclusion. In the words of the late Sir Fred Hoyle:
A common sense interpretation of the facts suggests that a superintellect has monkeyed with physics, as well as with chemistry and biology, and that there are no blind forces worth speaking about in nature. The numbers one calculates from the facts seem to me so overwhelming as to put this conclusion almost beyond question.
(The Universe: Past and Present Reflections, Engineering and Science, November 1981, p. 12.)
 |