OK, let’s start with a very simple fact: eukaryotic genes have introns.
IOWs, they are not continuous. They are made of exons and introns: exon – intron – exon – intron – exon and so on. Exons code for the protein. Introns don’t.
So, when the content of the gene is copied to the mRNA, introns must be cut away, and only exons are retained, in order to be translated, so that the mature mRNA can be transferred to the cytoplasm and translated by the ribosome.
This process of removing introns is called splicing.
Now, a few clarifications:
a) Introns exist in prokaryotes too, but they are rather rare. For our purposes, we will only discuss introns in eukaryotes.
b) Introns exist in many different types of genes. For our purposes, we will discuss only those in protein coding genes.
c) The origin and possible function of introns is, still, a mystery.
d) Introns are usually longer than exons. In humans, for example, they amount to approximately 35% of the whole geneome, vs about 1.5% of coding exons.
e) However, the amount and length of introns can vary a lot in different organims. An extreme example is yeast (s. cerevisiae), whose genome contains a very small amount of introns (about 250 out of about 6250 genes).
When the gene if transcribed, both exons and introns are transcribed. A 5′ UTR (Untranslated region) and 3′ UTR, is also part of the pre-mRNA.
Fig. 1
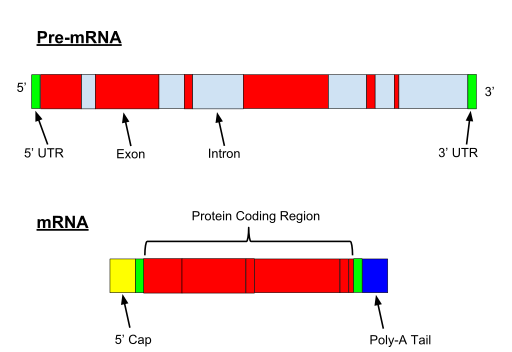
So the question is: how are introns removed from pre-mRNA? IOWs, how is splicing achieved?
And there is more. As everybody probably knows, splicing is not always done in the same way. Different isoforms of the same protein can be obtained by alternative splicing, and they can have functional differences. I will not go into details about that, but here is the Wikipedia page about alternative splicing:
https://en.wikipedia.org/wiki/Alternative_splicing
So, how is splicing done, and how is alternative splicing regulated?
We know much about the first question, very little about the second.
There are three ways to perform splicing:
- Spliceosomal splicing
- Self-splicing
- tRNA splicing
The last two modalities are rare, and can be found both in prokaryotes and eukaryotes. I will not discuss them here.
So, the subject of this OP is spliceosomal splicing, which is restricted to eukaryotes.
Moreover, I will discuss only the major spliceosome, which is responsible for the vast majority of splicing in eukaryotes. It must be said, however, that also a minor spliceosome exists, and that it acts in a minority of cases.
So, the spliceosome.
The first important point is:
It is an amazing molecular machine. Even more, it is an amazing molecular cycle, involving many different stages each of which is an amazing molecular machine.
Let’s see. Here is a figure which summarizes the main stages of the spliceosome cycle:
Fig. 2
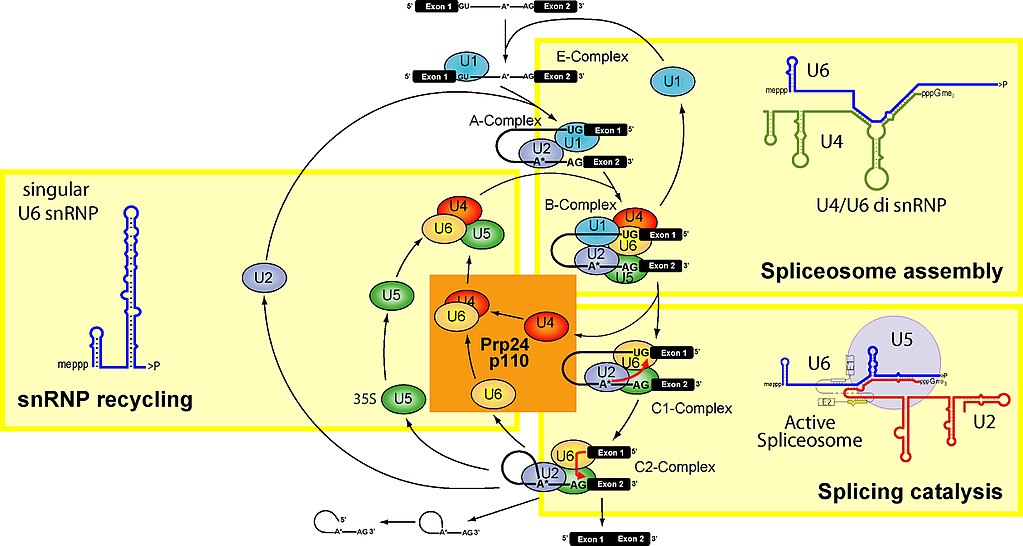
To make it simple, the spliceosome units are built upon 5 specific RNAs, called small nuclear RNAs (snRNA). These are, in humans:
U1 (164 bases), U2 (187 bases), U4 (145 bases), U5 (116 bases), U6 (107 bases)
They are transcribed from multiple gene copies. While their sequences are not particularly conserved (U6 being the most conserved of all), their secondary structure seems to be very conserved.
snRNAs are very important in the spliceosome, because they seem to be responsible for the catalytic activities.
Each of the 5 snRNAs forms a complex with proteins, and the complex takes the name of snRNP. The whole spliceosome includes at least 145 different proteins, maybe more, some of which are still not well known.
I will mention here some of the most important:
U1 snRNP:
U1 snRNP 70 kDa (P08621, 437 AAs)
U1 snRNP A (P09012, 282 AAs)
U1 snRNP C (P09234, 159 AAs)
Sm proteins: 7 small proteins (76 – 240 AAs) which form the “Sm core” ring in spliceosome subunits U1, U2, U4, U5, a ring which hosts the specific snRNA molecule.
U2 snRNP:
U2 snRNP A’ (P09661, 255 AAs)
U2 snRNP B” (P08579, 225 AAs)
SF3a120 (Q15459, 793 AAs)
SF3a66 (Q15428, 464 AAs)
SF3a60 (Q12874, 501 AAs)
SF3b155 (O75533, 1304 AAs)
SF3b145 (Q13435, 895 AAs)
SF3b130 (Q15393, 1217 AAs)
SF3b49 (Q15427, 424 AAs)
SF3b14a/p14 (Q9Y3B4, 125 AAs)
SF3b10 (Q9BWJ5, 86 AAs)
Sm proteins
U4/U6 snRNP:
Prp3 (O43395, 683 AAs)
Prp31 (Q8WWY3, 499 AAs)
Prp4 (O43172, 522 AAs)
Cyph ( O43447, 177 AAs)
15.5 K (P55769, 128 AAs)
Sm proteins (for the U4 snRNA)
Lsm proteins: a number of proteins similar to Sm proteins (usually Lsm 2-8), which form a specific ring for the U6 snRNA.
U5 snRNP:
Prp8 (Q6P2Q9, 2335 AAs)
BRR2 (O75643, 2136 AAs)
Snu114 (Q15029, 972 AAs)
Prp6 (O94906, 941 AAs)
Prp28 (Q9BUQ8, 820 AAs)
52 K (O95400, 341 AAs)
40 K (Q96DI7, 357 AAs)
Sm proteins
Additional proteins in the U4/U6/U5 complex:
hSnu66 (O43290, 800 AAs)
hSad1 (Q53GS9, 565 AAs)
27 K (Q8WVK2, 155 AAs)
OK, these are only the main components, and the best understood. We are still far from the sum total of 145/150 proteins which are involved in the spliceosome cycle.
But how does it work?
Always in brief. Here is a typical exon-intron structure:
Fig. 3
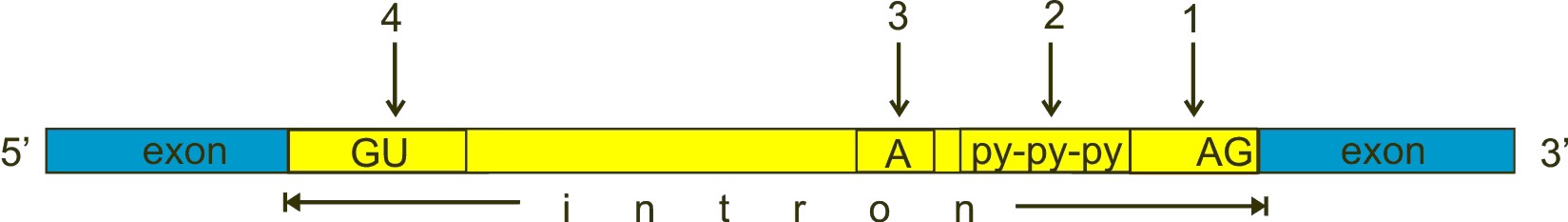
GU, A and AG are nucleotides almost universally conserved in all introns, approximately at the positions shown in the figure, and which have a fundamental role in splicing. However, the real stuff is much more complex than this (see the splicing code section). GU (n. 4 in the figure) is near to the 5′ end of the intron, AG (n. 1) near to the 3′ end. The A (n. 3 in the figure) is called “the branch point”. The py-py-py (n. 2 in the figure) is the “polypyrimidine tract”.
a) The U1 subunit binds to the GU sequence at the 5′ splice site in the intron
b) The U2 subunit binds to the “branch point”.
c) The U4/U6/U5 binds to the complex.
d) Numeorus further modifications take place, causing the formation of a “lariat” (including the intron), which is then cleaved, while the two exons are ligated.
I will spare you the many complexities in all the various steps, which are well summarized (in a very simplified way) in this Wikipedia page:
See in particular the “Formation and activity” section.
Or, if you like more detail, here:
Spliceosome Structure and Function
And here is a very good video on the whole splicing process in yeast:
Now, if somebody still has doubts about the complexity of this molecular machine/process, let’s consider some important aspects.
- 1. The spliceosome is a molecular machine which appears in eukaryotes.
I quote from this paper (in the abstract):
Origin and evolution of spliceosomal introns
There is no indication that any prokaryote has ever possessed a spliceosome or introns in protein-coding genes, other than relatively rare mobile self-splicing introns.
The following Table shows how some of the main proteins involved in the spliceosome activity show practically no trace of homology in prokaryotes. I have included also included in the table two examples which show low homologies due to some domain which is already expressed in prokaryotes: Prp4, whose 209 bits of homology are due to a specific domain, WD40, and Prp28, which exhibits 313 bits linked to the DEXDc domain. The point is: many of the spliceosome proteins are complete novelties in eukaryotes, but not all of them.
| Protein | Bacteria | Archea |
| U1 snRNP 70 kDa (P08621, 437 AAs) | 67 | – |
| U2 SF3b130 (Q15393, 1217 AAs) | 43.5 | – |
| U4/U6 hPrp3 (O43395, 683 AAs) | – | – |
| U4/U6/U5 hSnu66 (O43290, 800 AAs) | – | – |
| U5 Prp8 (Q6P2Q9, 2335 AAs) | – | – |
| U5 Prp6 (O94906, 941 AAs) | 100 | – |
| U4/U6 hPrp4 (O43172, 522 AAs) | 209 | 150 |
| U/5 Prp28 (Q9BUQ8, 820 AAs) | 313 | 286 |
I will analyze in more detail one of the most important proteins in the spliceosome, Prp8, in the last part of this OP
(Just a technical note: if you blast Prp8, you will find 3 hits which are obviously an error due to unverified sequences, probably cases of contamination).
- 2. The spliceosome is a molecular machine which is universally present in eukaryotes.
All eukaryotes have introns, even if in very different degrees, and the spliceosome, even if in some organisms parts of the spliceosome complex can be lost. For a more detailed discussion, look at the Rogozin paper quoted above (Origin and evolution of spliceosomal introns), in particular the section:
Intron density, size and distribution in protein coding genes across the eukaryote domain
I quote this important conclusion:
As pointed out above, despite the existence of numerous, diverse intron-poor genomes, eukaryotes do not lose the “last” intron or the spliceosome although degradation of the spliceosome including loss of many components does occur, e.g. in yeast. The only firmly established exception is the tiny genome of a nucleomorph (an extremely degraded intracellular symbiont of algae) that has lost both all the introns and the spliceosome [7]; preliminary genomic data indicate that all introns might have been lost also in a microsporidium, a highly degraded intracellular parasite distantly related to Fungi [54].
So, whe can conclude that both introns and the spliceosome are a universal feature of eukaryotes, the few exceptions being simply cases of loss of information.
- 3. The spliceosome is a molecular machine whose information is extremely conserved throughout eukaryotes, up to humans.
I have already mentioned that the 5 RNAs which form the core of the spliceosome are not extremely conserved at sequence level, even if they are extremely conserved at structure level.
However, many of the proteins that compose the spliceosome show an amazing sequence conservation throughout eukaryotes. Now, even if we cannot be certain of when eukaryoyes really emerged, and of their early evolutionary history (both issues being at present highly controversial), we can reasonably assume that protein sequences which are highly conserved in all eukaryotes have been conserved for something like 2 billion years (more or less). As anybody who has followed my previous OPs about information conservation in vertebrates certainly knows, that is an evolutionary time frame which certainly allows us to equal conservation to extremely high functional constraint.
But how conserved are spliceosome proteins? We can analyze a few of them with my usual methodology: looking at human conserved information. The results shows that many proteins involved in the spliceosome are amazingly conserved in all eukaryotes. While there are a few cases which have a rather different evolutionary history, this is by far the most common behaviour for spliceosomal proteins.
Here is a sample of some important sequences that show high homology with the human form in all major groups of single celled eukaryotes. The 5 groups of single celled organisms chosen here, indeed, cover rather well the whole range of single celled eukaryotes.
Fig. 4
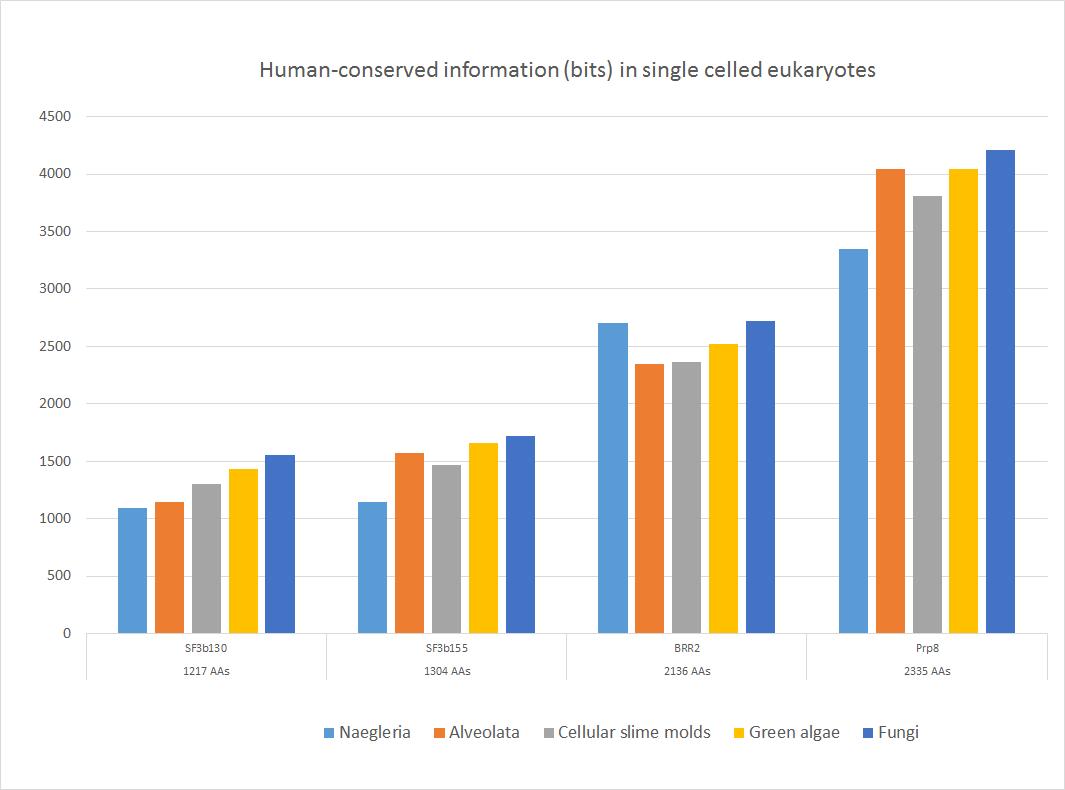
These four important proteins, as shown, have an amazing amount of information shared with the human form, ranging from more than 1000 to more than 4000 bits. In bits per AA, the range goes from 0.88 to 1.80 bits per aminoacid (baa). As can be seen, the highest homology is found in fungi, as expected, because fungi are the most likely ancestors of metazoa. The lowest homologies are observed in Naegleria (Excavata) or in Alveolata.
Of course, these proteins remain highly similar to the human form in the following evolutionary history in Metazoa.
So, we can safely state that most spliceosomal proteins, while emerging almost entirely in eukaryotes and showing only trivial homologies with prokaryotes, were probably already universally present in the Last Universal Eukaryotic Ancestor (LECA), and in a form already very similar at sequence level to what we observe in metazoa and in humans.
- 4. The spliceosome is a wonderful example of irreducibly complexity.
OK, we have already said that splicing can be achieved in at least three different ways. For example, bacterial introns, although rare, are of the self-splicing type. So, we know that the generic function of splicing introns can be implemented in different ways.
But eukaryotic introns are of the spliceosomal type, and they are spliced only by the spliceosome.
We have also said that a minor spliceosome also exists. It shares some featues with the major spliceosome, but it is a different structure and acts on different, and much rarer, introns.
So, for the vast majority of eukaryotic introns, the major spliceosome, and only the major spliceosome, can effectively accomplish the splicing.
Now, I don’t mean here that the major spliceosome must always be absolutely complete, with all its 150 proteins, to be able to work. That’s not what I mean when I say that it is irreducibly complex.
Maybe in some organisms the spliceosome can be partially defective, and still work. It is difficult to say, because we still don’t understand the role of all the components of the spliceosome.
But however, as far as we can understand, most of the principal features must be present, because, as we have seen, the splicing is the result of a complex cycle, involving the RNAs and the subunits, and all stages are essential to the final result.
So, the spliceosome is certainly highly irreducibly complex, even if we may not be able to clearly identify the essential nucleus of molecules which is absolutely necessary to the minimal function.
Moreover, the spliceosome would be useless if spliceosomal introns did not exist, with their properties and code (see later), and spliceosomal introns could not exist if the spliceosome were not there to splice them, because otherwise transcription would be completely ineffective. So, in that sense, spliceosomal introns and the spliceosome are a good example of chicken egg paradox, or we could also say that they form an irreducibly complex system at a higher level. And remember, the whole system seems to have been already present, very much similar to its current form in humans, in LECA, as we have seen.
But, of course, our darwinist friends will simply say that they have co-evolved! 🙂
Moreover, we can and should ask ourselves: why is the spliceosome so complex? The answer is not easy, because we still understand very little, but it is certainly related to the complexity of the splicing code, and to the fundamental issue of alternative splicing.
The Splicing code.
To splice introns by localizing 5 (or a few more) conserved nucleotides and then cutting at the ends of a lariat and rejoining the two exons is certainly a complex task, but apparently not so complex that one of the biggest and most impressing known molecular machines is needed for that. But the simple truth is that recognizing and appropriately splicing all introns is a much more complex task than that.
That is due to the simple fact that conserved nucleotides at the ends are not a sufficient signal to identify the segment that has to be spliced, and that a lot of other components (not always well understood) are necessary to that, and that the splicing is not made always in the same way, and that alternative splicing is a very powerful tool for transcription regulation.
The subject is very complex, and I will not deal with it in depth. However, Those interested could look at this recent review:
Unfortunately, the paper is paywalled, but the abstract is very informative.
The complexity of the splicing code can give us some insight about the true reasons for the complexity of the spliceosome, and definitely supposrts the idea that the whole system, introns, splicing code and spliceosome, is irreducibly complex.
The Prp8 protein.
I will add a few words about this proteins, which is probably the most amazing component of the spliceosome, and well represents its essential features.
This protein has many amazing charactertistics:
a) It is, as far as I can say, the longest protein in the whole spliceosomal system, and a very long protein indeed: 2335 AAs.
b) It is completely absent in prokaryotes.
c) It is extremely conserved in eukaryotes, probably the most conserved protein in the whole spliceosomal complex (see Fig. 4). In Naegleria, we have 3345 bits of homology with the human protein, corresponding to 1.4 baa, while in fungi we have the amazing result of 4211 bits of human-conserved information, corresponding to 1.8 baa. IOWs, in fungi we already find almost 90% of the functional information present in human Prp8 (remember, the highest possible bitscore is about 1.2 baas), corresponding to 1995 identities (86%) and 2155 positives (92%): a result which is incredibly rare, considering that such information has been conserved for about 2 billion years.
Just to emphasize the importance of this fact, here is the blast between the human protein and the best hit in fungi (Basidiobolus meristosporus):

d) It is a protein which is extremely important for the function of the spliceosome. Here is a very good paper about this protein and its functional relevance:
Prp8 protein: At the heart of the spliceosome
I quote from the conclusions:
Prp8p is central to the expression of all nuclear intron-containing mRNAs. In higher eukaryotes, it is responsible for processing thousands of transcripts in alternative splicing pathways, and in both U2 and U12 spliceosomes. It is important for the pathology of human disease, as all eukaryotic pathogens and parasites require Prp8p to functionally express their genes, in some cases via the trans-spliceosome. Retinitis Pigmentosa, a human genetic disorder that causes progressive blindness, positions Prp8p as a target for therapeutic medicine.
Moreover, another way to assess the functional constraints of a protein is to check its tolerance to polymorphisms in humans. That can be done consulting a very recent and useful database, the ExAC browser, which reports data from about 60000 human genomes.
ExAC gives two important metrics to assess how much a protein is tolerant to polymorphisms and variants. I quote here from the site FAQ:
What are the constraint metrics?
For synonymous and missense, we created a signed Z score for the deviation of observed counts from the expected number. Positive Z scores indicate increased constraint (intolerance to variation) and therefore that the gene had fewer variants than expected. Negative Z scores are given to genes that had a more variants than expected.
For LoF, we assume that there are three classes of genes with respect to tolerance to LoF variation: null (where LoF variation is completely tolerated), recessive (where heterozygous LoFs are tolerated), and haploinsufficient (where heterozygous LoFs are not tolerated). We used the observed and expected variants counts to determine the probability that a given gene is extremely intolerant of loss-of-function variation (falls into the third category). The closer pLI is to one, the more LoF intolerant the gene appears to be. We consider pLI >= 0.9 as an extremely LoF intolerant set of genes.
Now, for Prp8 we get the following results:
| Constraint from ExAC |
Expected no. variants |
Observed no. variants |
Constraint Metric |
|---|---|---|---|
| Synonymous | 366.6 | 460 | z = -3.02 |
| Missense | 920.2 | 266 | z = 10.55 |
| LoF | 92.6 | 8 | pLI = 1.00 |
That means the following:
a) The number of observed missense variants is so low vs expected (266 vs 920.2) that the z value is 10.55 (IOWs, 10.55 standard deviations, more than 10 sigma). Believe me, this is a really exceptional result, most proteins are much more tolerant to missense variants.
b) The probability of loss of function is 1. That means that even heterozygous LoF is absolutely not tolerated.
This is an extremely functional molecule, if ever there was one!
A brief conclusion.
To sum up the meaning of this rather long discussion, I would simply say:
- The introns – spliceosome system is a molecular machine of amazing complexity. I have touched only its main aspects in this OP, but believe me, there are layers of complexity there that would require a whole treatise and that I have not even started to mention here.
- Whoever can really believe that all this can be explained by some RV + NS model is, IMO, really admirable for his faith in a wrong paradigm.