Sometimes, the very dismissiveness of hyperskeptical objections is their undoing, as in this case from TSZ:
Pesky EleP(T|H)ant
Over at Uncommon Descent KirosFocus repeats the same old bignum arguments as always. He seems to enjoy the ‘needle in a haystack’ metaphor, but I’d like to counter by asking how does he know he’s not searching for a needle in a needle stack? . . .
What had happened, is that on June 24th, I had posted a discussion here at UD on what Functionally Specific Complex Organisation and associated Information (FSCO/I) is about, including this summary infographic:
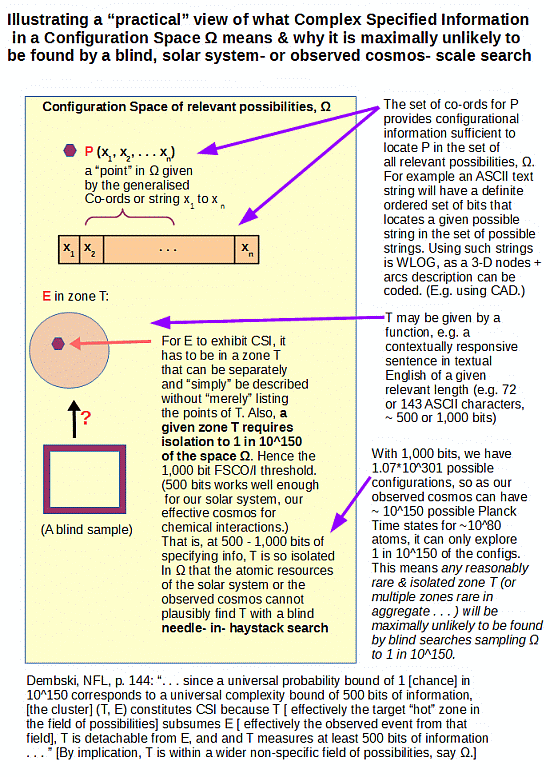 Instead of addressing what this actually does, RTH of TSZ sought to strawmannise and rhetorically dismiss it by an allusion to the 2005 Dembski expression for Complex Specified Information, CSI:
Instead of addressing what this actually does, RTH of TSZ sought to strawmannise and rhetorically dismiss it by an allusion to the 2005 Dembski expression for Complex Specified Information, CSI:
χ = – log2[10^120 ·ϕS(T)·P(T|H)].
–> χ is “chi” and ϕ is “phi” (where, CSI exists if Chi > ~ 1)
. . . failing to understand — as did the sock-puppet Mathgrrrl [not to be confused with the Calculus prof who uses that improperly appropriated handle) — that by simply moving forward to the extraction of the information and threshold terms involved, this expression reduces as follows:
To simplify and build a more “practical” mathematical model, we note that information theory researchers Shannon and Hartley showed us how to measure information by changing probability into a log measure that allows pieces of information to add up naturally:
Ip = – log p, in bits if the base is 2. That is where the now familiar unit, the bit, comes from. Where we may observe from say — as just one of many examples of a standard result — Principles of Comm Systems, 2nd edn, Taub and Schilling (McGraw Hill, 1986), p. 512, Sect. 13.2:
Let us consider a communication system in which the allowable messages are m1, m2, . . ., with probabilities of occurrence p1, p2, . . . . Of course p1 + p2 + . . . = 1. Let the transmitter select message mk of probability pk; let us further assume that the receiver has correctly identified the message [[–> My nb: i.e. the a posteriori probability in my online discussion here is 1]. Then we shall say, by way of definition of the term information, that the system has communicated an amount of information Ik given by
I_k = (def) log_2 1/p_k (13.2-1)
xxi: So, since 10^120 ~ 2^398, we may “boil down” the Dembski metric using some algebra — i.e. substituting and simplifying the three terms in order — as log(p*q*r) = log(p) + log(q ) + log(r) and log(1/p) = – log (p):
Chi = – log2(2^398 * D2 * p), in bits, and where also D2 = ϕS(T)
Chi = Ip – (398 + K2), where now: log2 (D2 ) = K2That is, chi is a metric of bits from a zone of interest, beyond a threshold of “sufficient complexity to not plausibly be the result of chance,” (398 + K2). So,
(a) since (398 + K2) tends to at most 500 bits on the gamut of our solar system [[our practical universe, for chemical interactions! ( . . . if you want , 1,000 bits would be a limit for the observable cosmos)] and(b) as we can define and introduce a dummy variable for specificity, S, where
(c) S = 1 or 0 according as the observed configuration, E, is on objective analysis specific to a narrow and independently describable zone of interest, T:Chi = Ip*S – 500, in bits beyond a “complex enough” threshold
- NB: If S = 0, this locks us at Chi = – 500; and, if Ip is less than 500 bits, Chi will be negative even if S is positive.
- E.g.: a string of 501 coins tossed at random will have S = 0, but if the coins are arranged to spell out a message in English using the ASCII code [[notice independent specification of a narrow zone of possible configurations, T], Chi will — unsurprisingly — be positive.
- Following the logic of the per aspect necessity vs chance vs design causal factor explanatory filter, the default value of S is 0, i.e. it is assumed that blind chance and/or mechanical necessity are adequate to explain a phenomenon of interest.
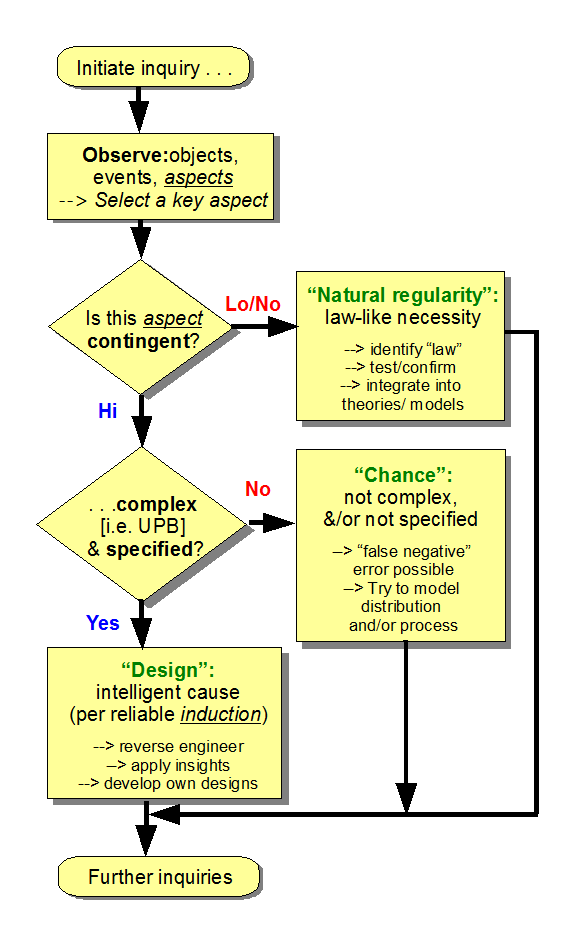
- S goes to 1 when we have objective grounds — to be explained case by case — to assign that value.
- That is, we need to justify why we think the observed cases E come from a narrow zone of interest, T, that is independently describable, not just a list of members E1, E2, E3 . . . ; in short, we must have a reasonable criterion that allows us to build or recognise cases Ei from T, without resorting to an arbitrary list.
- A string at random is a list with one member, but if we pick it as a password, it is now a zone with one member. (Where also, a lottery, is a sort of inverse password game where we pay for the privilege; and where the complexity has to be carefully managed to make it winnable. )
- An obvious example of such a zone T, is code symbol strings of a given length that work in a programme or communicate meaningful statements in a language based on its grammar, vocabulary etc. This paragraph is a case in point, which can be contrasted with typical random strings ( . . . 68gsdesnmyw . . . ) or repetitive ones ( . . . ftftftft . . . ); where we can also see by this case how such a case can enfold random and repetitive sub-strings.
- Arguably — and of course this is hotly disputed — DNA protein and regulatory codes are another. Design theorists argue that the only observed adequate cause for such is a process of intelligently directed configuration, i.e. of design, so we are justified in taking such a case as a reliable sign of such a cause having been at work. (Thus, the sign then counts as evidence pointing to a perhaps otherwise unknown designer having been at work.)
- So also, to overthrow the design inference, a valid counter example would be needed, a case where blind mechanical necessity and/or blind chance produces such functionally specific, complex information. (Points xiv – xvi above outline why that will be hard indeed to come up with. There are literally billions of cases where FSCI is observed to come from design.)
xxii: So, we have some reason to suggest that if something, E, is based on specific information describable in a way that does not just quote E and requires at least 500 specific bits to store the specific information, then the most reasonable explanation for the cause of E is that it was designed. The metric may be directly applied to biological cases:
Using Durston’s Fits values — functionally specific bits — from his Table 1, to quantify I, so also accepting functionality on specific sequences as showing specificity giving S = 1, we may apply the simplified Chi_500 metric of bits beyond the threshold:
RecA: 242 AA, 832 fits, Chi: 332 bits beyondSecY: 342 AA, 688 fits, Chi: 188 bits beyondCorona S2: 445 AA, 1285 fits, Chi: 785 bits beyond
Where, of course, there are many well known ways to obtain the information content of an entity, which automatically addresses the “how do you evaluate p(T|H)” issue. (As has been repeatedly pointed out, just insistently ignored in the rhetorical intent to seize upon a dismissive talking point.)
There is no elephant in the room.
Apart from . . . the usual one design objectors generally refuse to address, selective hyperskepticism.
But also, RTH imagines there is a whole field of needles, refusing to accept that many relevant complex entities are critically dependent on having the right parts, correctly arranged, coupled and organised in order to function.
That is, there are indeed empirically and analytically well founded narrow zones of functional configs in the space of possible configs. By far and away most of the ways in which the parts of a watch may be arranged — even leaving off the ever so many more ways they can be scattered across a planet or solar system– will not work.
The reality of narrow and recognisable zones T in large spaces W beyond the blind sampling capacity — that’s yet another concern — of a solar system of 10^57 atoms or an observed cosmos of 10^80 or so atoms and 10^17 s or so duration, is patent. (And if RTH wishes to dismiss this, let him show us observed cases of life spontaneously organising itself out of reasonable components, say soup cans. Or, of watches created by shaking parts in drums, or of recognisable English text strings of at least 72 characters being created through random text generation . . . which last is a simple case that is WLOG, as the infographic points out. As, 3D functional arrangements can be reduced to code strings, per AutoCAD etc.)
Finally, when the material issue is sampling, we do not need to generate grand probability calculations.

For, once we are reasonably confident that we are looking at deeply isolated zones in a field of possibilities, it is simple to show that unless a “search” is so “biased” as to be decidedly not random and decidedly not blind, only a blind sample on a scope sufficient to make it reasonably likely to catch zones T in the field W would be a plausible blind chance + mechanical necessity causal account.
But, 500 – 1,000 bits (a rather conservative threshold relative to what we see in just the genomes of life forms) of FSCO/I is (as the infographic shows) far more than enough to demolish that hope. For 500 bits, one can see that to give every one of the 10^57 atoms of our solar system a tray of 500 H/T coins tossed and inspected every 10^-14 s — a fast ionic reaction rate — would sample as one straw to a cubical haystack 1,000 LY across, about as thick as our galaxy’s central bulge. If such a haystack were superposed on our galactic neighbourhood and we were to take a blind, reasonably random one-straw sized sample it would with maximum likelihood be straw.
As in, empirically impossible, or if you insist, all but impossible.
It seems that objectors to design inferences on FSCO/I have been reduced to clutching at straws. END