I have recently commented on another thread:
about a paper that (very correctly) describes cells as dynamic, far from equilibrium systems, rather than as “traditional” machines.
That is true. But, of course, the cell implements the same functions as complex machines do, and much more. My simple point is that, to do that, you need much greater functional complexity than you need to realize a conventional machine.
IOWs, dynamic, far from equilibrium systems that can be as successful as a conventional machine, or more, must certainly be incredibly complex and amazing systems, systems that defy everything else that we already know and that we can conceive. They must not only implement their functional purposes, but they must do that by “harnessing” the constantly changing waves of change, of random noise, of improbability. I have commented on those ideas in the mentioned thread, at posts #5 and #8, and I have quoted at posts #11 and #12 a couple of interesting and pertinent papers, introducing the important concept of robustness: the ability to achieve reliable functional results in spite of random noise and disturbing variation.
In this OP, I would like to present in some detail a very interesting system that shows very well what we can understand, at present, of that kind of amazing systems.
The system I will discuss here is an old friend: it is the NF-kB system of transcription factors (nuclear factor kappa-light-chain-enhancer of activated B cells). We are speaking, therefore, of transcription regulation, a very complex topic that I have already discussed in some depth here:
I will remind here briefly that transcription regulation is the very complex process that allows cells to be completely different using the same genomic information: IOWs, each type of cell “reads” differently the genes in the common genome, and that allows the different types of cell differentiation and the different cell responses in the same cell type.
Transcription regulation relies on many different levels of control, that are summarized in the above quoted OP, but a key role is certainly played by Transcription Factors (TFs), proteins that bind DNA and act as activators or inhibitors of transcription at specific sites.
TFs are a fascinating class of proteins. There are a lot of them (1600 – 2000 in humans, almost 10% of all proteins), and they are usually medium sized proteins, about 500 AA long, containing at least one highly conserved domain, the DNA binding domain (DBD), and other, often less understood, functional components.
I quote again here a recent review about human TFs:
The Human Transcription Factors
The NK-kB system is a system of TFs. I have discussed it in some detail in the discussion following the Ubiquitin thread, but I will describe it in a more systematic way here.
In general, I will refer a lot to this very recent paper about it:
Considering Abundance, Affinity, and Binding Site Availability in the NF-kB Target Selection Puzzle
The NF-kB system relies essentially on 5 different TFs (see Fig. 1 A in the paper):
- RelA (551 AAs)
- RelB (579 AAs)
- c-Rel (619 AAs)
- p105/p50
(968 AAs) - p100/p52 (900 AAs)
Those 5 TFs work forming dimers, homodimers or heterodimers, for a total of 15 possible compbinations, all of which have been found to work in the cell, even if some of them are much more common.
Then there are at least 4 inhibitor proteins, collectively called IkBs.
The mechanism is apparently simple enough. The dimers are inhibited by IkBs and therefore they remain in the cytoplasm in inactive form.
When an appropriate signal arrives to the cell and is received by a membrane receptor, the inhibitor (the IkB molecule) is phosphorylated and then ubiquinated and detached from the complex. This is done by a protein complex called IKK. The free dimer can then migrate to the nucleus and localize there, where it can act as a TF, binding DNA.
This is the canonical activation pathway, summarized in Fig. 1. There is also a non canonical activation pathway, that we will not discuss for the moment.
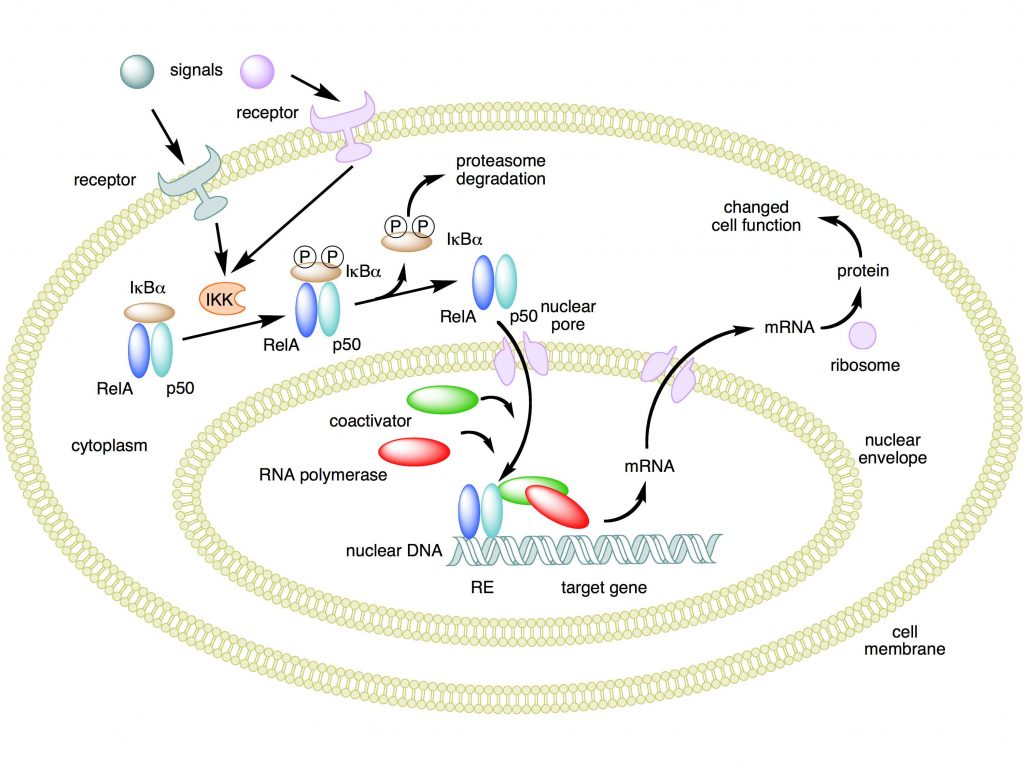
Mechanism of NF-κB action. In this figure, the NF-κB heterodimer consisting of Rel and p50 proteins is used as an example. While in an inactivated state, NF-κB is located in the cytosol complexed with the inhibitory protein IκBα. Through the intermediacy of integral membrane receptors, a variety of extracellular signals can activate the enzyme IκB kinase (IKK). IKK, in turn, phosphorylates the IκBα protein, which results in ubiquitination, dissociation of IκBα from NF-κB, and eventual degradation of IκBα by the proteasome. The activated NF-κB is then translocated into the nucleus where it binds to specific sequences of DNA called response elements (RE). The DNA/NF-κB complex then recruits other proteins such as coactivators and RNA polymerase, which transcribe downstream DNA into mRNA. In turn, mRNA is translated into protein, resulting in a change of cell function.
Attribution: Boghog2 at English Wikipedia [Public domain]
Now, the purpose of this OP is to show, in greater detail, how this mechanism, apparently moderately simple, is indeed extremely complex and dynamic. Let’s see.
The stimuli.
First of all, we must understand what are the stimuli that, arriving to the cell membrane, are capable to activate the NF-kB system. IOWs, what are the signals that work as inputs.
The main concept is: the NF-kB system is a central pathway activated by many stimuli:
- Inflammation
- Stress
- Free
radicals - Infections
- Radiation
- Immune
stimulation
IOWs, a wide variety of aggressive stimuli can activate the system
The extracellular signal arrives to the cell usually through specific cytokines, for example TNF, IL1, or through pathogen associated molecules, like bacterial lipopolysaccharides (LPS). Of course there are different and specific membrane receptors, in particular IL-1R (for IL1) , TNF-R (for TNF), and many TLRs (Toll like receptors, for pathogen associated structures). A special kind of activation is implemented, in B and T lymphocytes, by the immune activation of the specific receptors for antigen epitopes (B cell receptor, BCR, and T cell receptor, TCR).
The process through which the activated receptor can activate the NF-kB dimer is rather complex: it involves, in the canonical pathway, a macromolecular complex called IKK (IkB kinase) complex, comprising two catalytic kinase subunits (IKKa and IKKb) and a regulatory protein (IKKg/NEMO), and involving in multiple and complex ways the ubiquitin system. The non canonical pathway is a variation of that. Finally, a specific protein complex (CBM complex or CBM signalosome) mediates the transmission from the immune BCR or TCR to the canonical pathway. See Fig. 2:
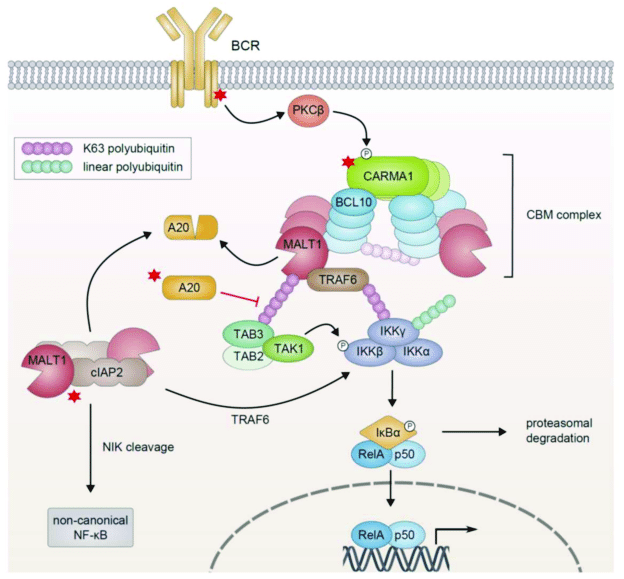
Figure 3 – NF-κB Activation in Lymphoid Malignancies: Genetics, Signaling, and Targeted Therapy
available via license: Creative Commons Attribution 4.0 International
I will not go into further details about this part, but those interested can have a look at this very good paper:
TLR-4, IL-1R and TNF-R signaling to NF-kB: variations on a common theme
In particular, Figg. 1, 2, 3.
In the end, as a result of the activation process, the IkB inhibitor is degraded by the ubiquitin system, and the NK-kB dimer is free to migrate to the nucleus.
An important concept is that this is a “rapid-acting” response system, because the dimers are already present, in inactive form, in the cytoplasm, and must not be synthesized de novo: so the system is ready to respond to the activating signal.
The response.
But what is the cellular response?
Again, there are multiple and complex possible responses.
Essentially, this system is a major regulator of innate and adaptive immune responses. As such, it has a central role in the regulation of inflammation, in immunity, in autoimmune processes, and in cancer.
Moreover, the NF-kB system is rather ubiquitous, and is present and active in many different cell types. And, as we have seen, it can be activated by different stimuli, in different ways.
So, the important point is that the response to activation must be (at least):
- Lineage-specific
- Stimulus-specific
IOWs, different cells must be able to respond differently, and each cell type must respond differently to different stimuli. That gives a wide range of possible gene expression patterns at the transcription level.
The following paper is a good review of the topic:
Selectivity of the NF-κB Response
For example, IL2 is induced by NF-kB activayion in T cells, but not in B cells (lineage specific response). Moreover, specific cell types can undergo specific, and often different, cell destinies after NF-kB activation: for example, NK-kB is strongly involved in the control and regulation of T and B cell development.
From:
30 years of NF-κB: a blossoming of relevance to human pathobiology
“B and T lymphocytes induce NF-κB in adaptive immune responses through the CARD11:Bcl10:MALT1 (CBM) complex (Hayden and Ghosh, 2008). Newly expressed genes promote lymphocyte proliferation and specific immune functions including antibody production by B cells and the generation of cytokines and other anti-pathogen responses by T cells.”
And, in the same cell type, certain promoters regulated by NF-kB require additional signaling (for example, in human dendritic cells promoters for Il6, Il12b, and MCP-1 require additional p38 histone phosphorylation to be activated), while others can be activated directly (stimulus-specific response).
So, to sum up:
- A variety of stimuli can activate the system in different ways
- The system itself has its complexity (different dimers)
- The response can be widely different, according to the cell type where it happens, and to the type of stimuli that have activated the system, and probably according to other complex variables.
- The possible responses include a wide range of regulations of inflammation, of the immune system, of cell specifications or modifications, and so on.
How does it work?
So, what do we know about the working of such a system?
I will ignore, for the moment, the many complexities of the activation pathways, both canonical and non canonical, the role of cyotkines and receptors and IKK complexes, the many facets of NEMO and of the involvement of the ubiquitin system.
For simplicity, we will start with the activated system: the IkB inhibitor has been released from the inactive complex in the cytoplasm, and some form of NF-kB dimer is ready to migrate to the nucleus.
Let’s remember that the purpose of this OP is to show that the system works as a dynamic, far from equilibrium system, rather than as a “traditional” machine. And that such a way to work is an even more amazing example of design and functional complexity.
To do that; I will rely mainly on the recent paper quoted at the beginning:
Considering Abundance, Affinity, and Binding Site Availability in the NF-kB Target Selection Puzzle
The paper is essentially about the NF-kB Target Selection Puzzle. IOWs, it tries to analyze what we know about the specificity of the response. How are specific patterns of transcription achieved after the activation of the system? What mechanisms allow the selection of the right genes to be transcribed (the targets) to implement the specific patterns according to cell type, context, and type of stimuli?
A “traditional” view of the system as a machine would try to establish rather fixed connections. For example, some type of dimer is connected to specific stimuli, and evokes specific gene patterns. Or some other components modulate the effect of NK-kB, generate diversification and specificity of the response.
Well, those ideas are not completely wrong. In a sense, the system does work also that way. Dimer specificity has a role. Other components have a role. In a sense, but only in a sense, the system works as though it were a traditional machine, and uses some of the mechanisms that we find in the concept of a traditional biological machine.
But that is only a tiny part of the real thing.
The real thing is that the system really works as a dynamic, far from equilibrium system, harnessing huge random/stochastic components to achieve robustness and complexity and flexibility of behavior in spite of all those non finalistic parts.
Let’s see how that happens, at least for the limited understanding we have of it. It is important to consider that this is a system that has been studied a lot, for decades, because of its central role in so many physiological and pathological contexts, and so we know many things. But still, our understanding is very limited, as you will see.
So, let’s go back to the paper. I will try to summarize as simply as possible the main concepts. Anyone who is really interested can refer to the paper itself.
Essentially, the paper analyzes three important and different aspects that contribute to the selection of targets at the genomic level by our TFs (IOWs, our NF-kB dimers, ready to migrate to the nucleus. As the title itself summarizes, they are:
- Abundance
- Affinity
- Binding site availability
1. Abundance
Abundance is referred here to two different variables: abundance of NF-kB Binding Sites in the genome and abundance of Nucleus-Localized NF-kB Dimers. Let’s consider them separately.
1a) Abundance of NF-kB Binding Sites in the genome:
It is well known that TFs bind specific sites in the genome. For NF-kB TFs, the following consensus kB site pattern has been found:
5′-GGGRNWYYCC-3′
where R, W, Y, and N, respectively denote purine, adenine or thymine, pyrimidine, and any nucleotide.
That simply means that any sequence corresponding to that pattern in the genome can, in principle, bind NF-kB dimers.
So the problem is: how many such sequences do exist in the human genome?
Well, a study based on RelA has evaluated about 10^4 consensus sequences in the whole genome, but as NF-kB dimers seem to bind even incomplete consensus sites, the total number of potential binding sites could be nearer to 10^6
1b) Abundance of Nucleus-Localized NF-kB Dimers:
An estimate of the abundance of dimers in the nucleus after activation of the system is that about 1.5 × 10^5 molecules can be found, but again that is derived from studies about RelA only. Moreover, the number of molecules and type of dimer can probably vary much according to cell type.
So, the crucial variable, that is the ratio between binding sites and available dimers, and which could help undertsand the rate of sites saturation in the nucleus, remains rather undecided, and it seems very likely that it can vary a lot in different circumstances.
But there is another very interesting aspect about the concentration of dimers in the nucleus. According to some studies, NF-kB seems to generate oscillations of its nuclear content in some cell types, and those oscillation can be a way to generate specific transcription patterns:
NF-kB oscillations translate into functionally related patterns of gene expression
For example, this very recent paper :
NF-κB Signaling in Macrophages: Dynamics, Crosstalk, and Signal Integration
shows at Fig. 3 the occupancy curve of binding sites at nuclear level after NF-kB activation in two different cell types.
In fibroblasts, the curve is a periodic oscillation, with a frequency that varies according to various factors, and translates into different transcription scenarios accordingly:
Gene expression dynamics scale with the period (g1) and amplitude (g2) of these oscillations, which are influenced by variables such as signal strength, duration, and receptor identity.
In macrophages, instead, the curve is rather:
a single, strong nuclear translocation event which persists for as long as the stimulus remains and tends to remain above baseline for an extended period of time.
In this case, the type of transcription will be probably regulated by the are under the curve, ratehr than by the period and amplitude of the oscialltions, as happened in fibroblasts.
Interestingly, while in previous studies it seemed that the concentration of nuclear dimers could be sufficient to saturate most or all binding sites, that has been found not to be the case in more recent studies. Again from the paper about abundance:
in fact, this lack of saturation of the system is necessary to generate stimulus- and cell-type specific gene expression profiles
Moreover, the binding itself seems to be rather short-lived:
Interestingly, it is now thought that most functional NF-kB interactions with chromatin—interactions that lead to a change in transcription—are fleeting… a subsequent study using FRAP in live cells expressing RelA-GFP showed that most RelA-DNA interactions are actually quite dynamic, with half-lives of a few seconds… Indeed, a recent study used single-molecule tracking of individual Halo-tagged RelA molecules in live cells to show that the majority (∼96%) of RelA undergoes short-lived interactions lasting on average ∼0.5 s, while just ∼4% of RelA molecules form more stable complexes with a lifetime of ∼4 s.
2. Affinity
Affinity of dimers for DNA sequences is not a clear cut matter. From the paper:
Biochemical DNA binding studies of a wide variety of 9–12 base-pair sequences have revealed that different NF-kB dimers bind far more sequences than previously thought, with different dimer species exhibiting specific but overlapping affinities for consensus and non-consensus kB site sequences.
IOWs, we have different dimers (15 different types) binding with varying affinity different DNA sequences (starting from the classical consensus sequence, but including also incomplete sequences). Remember that those sequences are rather short (the consensus sequence is 10 nucleotides long), and that there are thousands of such sequences in the genome.
Moreover, different bindings can affect transcription differently. Again, from the paper:
How might different consensus kB sites modulate the activity of the NF-kB dimers? Structure-function studies have shown that binding to different consensus kB sites can alter the conformation of the bound NF-kB dimers, thus dictating dimer function When an NF-kB dimer interacts with a DNA sequence, side chains of the amino acids located in the DNA-binding domains of dimers contact the bases exposed in the groove of the DNA. For different consensus kB site sequences different bases are exposed in this groove, and NF-kB seems to alter its conformation to maximize interactions with the DNA and maintain high binding affinity. Changes in conformation may in turn impact NF-kB binding to co-regulators of transcription, whether these are activating or inhibitory, to specify the strength and dynamics of the transcriptional response. These findings again highlight how the huge array of kB binding site sequences must play a key role in modulating the transcription of target genes.
Quite a complex scenario, I would say!
But there is more:
Finally, as an additional layer of dimer and sequence-specific regulation, each of the subunits can be phosphorylated at multiple sites with, depending on the site, effects on nearly every step of NF-kB activation.
IOWs, the 15 dimers we have mentioned can be phosphorylated in many different ways, and that changes their binding affinities and their effects on transcription.
This section of the paper ends with a very interesting statement:
Overall, when considering the various ways in which NF-kB dimer abundances and their affinity for DNA can be modulated, it becomes clear that with these multiple cascading effects, small differences in consensus kB site sequences and small a priori differences in interaction affinities can ultimately have a large impact on the transcriptional response to NF-kB pathway activation.
Emphasis mine.
This is interesting, because in some way it seems to suggest that the whole system acts like a chaotic system, at least at some basic level. IOWs, small initial differences, maybe even random noise, can potentially affect deeply the general working of the whole systems.
Unless, of course, there is some higher, powerful level of control.
3. Availability of high affinity kB binding sequences
We have seen that there is a great abundance and variety of binding sequences for NF-kB dimers in the human genome. But, of course, those sequences are not necessarily available. Different cell types will have a different scenario of binding sites availability.
Why?
Because, as we know, the genome and chromatin are a very dynamic system, that can exist in many different states, continuosly changing in different cell types and, in the same cell type, in different conditions..
We know rather well the many levels of control that affect DNA and chromatin state. In brief, they are essentially:
- DNA methylation
- Histone modifications (methylation, acetylation, etc)
- Chromatin modifications
- Higher levels of organization, including nuclear localization and TADs (Topologically Associating Domains)
For example, from the paper:
The promoter regions of early response genes have abundant histone acetylation or trimethylation prior to stimulation [e.g., H3K27ac, (67) and H4K20me3, (66)], a chromatin state “poised” for immediate activation… In contrast, promoters of late genes often have hypo-acetylated histones, requiring conformational changes to the chromatin to become accessible. They are therefore unable to recruit NF-kB for up to several hours after stimulation (68), due to the slow process of chromatin remodeling.
We must remember that each wave of NK-kB activation translates into the modified transcription of a lot of different genes at the genome level. It is therefore extremely important to consider what genes are available (IOWs, their promoters can be reached by the NF-kB signal) in each cell type and cell state.
The paper concludes:
Taken together, chromatin state and chromatin organization strongly influence the selection of DNA binding sites by NF-kB dimers and, most likely, the selection of the target genes that are regulated by these protein-DNA interaction events. Analyses that consider binding events in the context of three-dimensional nuclear organization and chromatin composition will be required to generate a more accurate view of the ways in which NF-kBDNA binding affects gene transcription.
This is the main scenario. But there are other components, that I have not considered in detail for the sake of brevity, for example competition between NF-kB dimers and the complex role and intervention of other co-regulators of transcription.
Does the system work?
But does the system work?
Of course it does. It is a central regulator, as we have said, of many extremely important biological processes, above all immunity. This is the system that decides how immune cells, T and B lymphocytes, have to behave, in terms of cell destiny and cell state. It is of huge relevance in all inflammatory responses, and in our defense against infections. It works, it works very well.
And what happens if it does not work properly?
Of course, like all very complex systems, errors can happen. Those interested can have a look at this recent paper:
30 years of NF-κB: a blossoming of relevance to human pathobiology
First of all, many serious genetic diseases have been linked to mutations in genes involved in the system. You can find a list in Table 1 of the above paper. Among them, for example, some forms of SCID, Severe combined immunodeficiency, one of the most severe genetic diseases of the immune system.
But, of course, a dysfunction of the NF-kB system has a very important role also in autoimmune diseases and in cancer.
Conclusions.
So, let’s try to sum up what we have seen here in the light of the original statement about biological systems that “are not machines”.
The NF-kB system is a perfect example. Even if we still understand very little of how it works, it is rather obvious that it is not a traditional machine.
A traditional machine would work differently. The signal would be transmitted from the membrane to the nucleus in the simplest possible way, without ambiguities and diversions. The Transcription Factor, once activated, would bind, at the level of the genome, very specific sites, each of them corresponding to a definite cascade of specific genes. The result would be clear cut, almost mechanical. Like a watch.
But that’s not the way things happen. There are myriads of variations, of ambiguities, of stochastic components.
The signal arrives to the membrane in multiple ways, very different one from the other: IL1, IL17, TNF, bacterial LPS, and immune activation of the B cell receptor (BCR) or the T cell receptor (TCR) are all possible signals.
The signal is translated to the NF-kB proteins in very different ways: canonical or non canonical activation, involving complex protein structures such as:
The CBM signalosome, intermediate between immune activation of BCR or TCR and canonical activation of the NF-kB. This complex is made of at least three proteins, CARD11, Bcl10 and MALT1.
The IKK complex in canonical activation: this is made of three proteins, IKK alpha, IKK beta, and NEMO. Its purpose is to phosphorylate the IkB, the inhibitor of the dimers, so that it can be ubiquinated and released from the dimer. Then the dimer can relocate to the nucleus.
Non canonical pathway: it involves the following phosphorylation cascade: NIK -> IKK alpha dimer -> Relb – p100 dimer -> Relb – p50 dimer (the final TF). It operates during the development of lymphoid organs and is responsible for the generation of B and T lymphocytes.
Different kinds of activated dimers relocate to the nucleus.
Different dimers, in varying abundance, interact with many different binding sites: complete or incomplete consensus sites, and probably others. The interaction is usually brief, and it can generate an oscillating pattern, or a more stable pattern
Completely different sets of genes are transcribed in different cell types and in different contexts, because of the interaction of NF-kB TFs with their promoters.
Many other factors and systems contribute to the final result.
The chromatin state of the cell at the moment of the NF-kB activation is essential to determine the accessibility of different binding sites, and therefore the final transcription pattern.
All these events and interactions are quick, unstable, far from equilibrium. A lot of possible random noise is involved.
In spite of that amazing complexity and potential stochastic nature of the system, reliable transcripion regulation and results are obtained in most cases. Those results are essential to immune cell differentiation, immune response, both innate and adaptive, inflammation, apoptosis, and many other crucial cellular processes.
So, let’s go back to our initial question.
Is this the working of a machine?
Of course it is! Because the results are purposeful, reasonably robust and reliable, and govern a lot of complex processes with remarkable elegance and efficiency.
But certainly, it is not a traditional machine. It is a lot more complex. It is a lot more beautiful and flexible.
It works with biological realities and not with transistors and switches. And biological realities are, by definition, far from equilibrium states, improbable forms of order that must continuously recreate themselves, fighting against the thermodynamic disorder and the intrinsic random noise that should apparently dominate any such scenario.
It is more similar to a set of extremely clever surfers who succeed in performing elegant and functional figures and motions in spite of the huge contrasting waves.
It is, from all points of view, amazing.
Now, Paley was absolutely right. No traditional machine, like a watch, could ever originate without design.
And if that is true of a watch, with its rather simple and fixed mechanisms, how much truer it must be for a system like NF-kB? Or, for that, like any cellular complex system?
Do you still have any doubts?
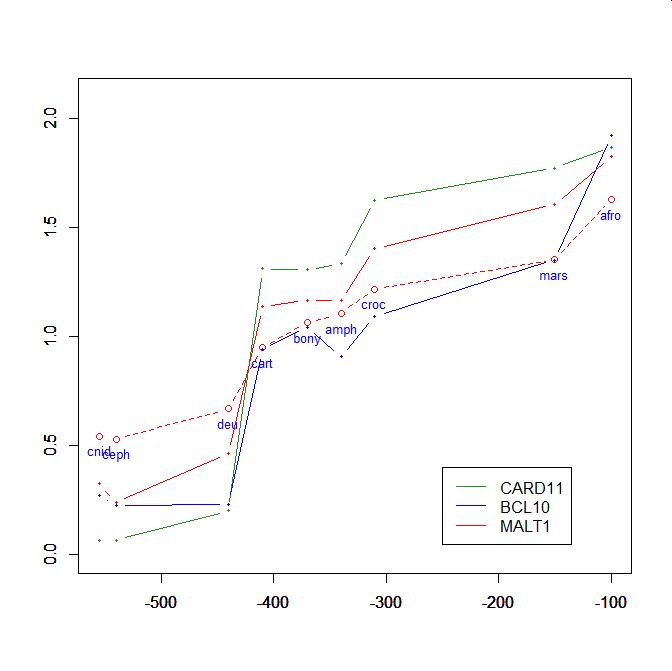
On the y axis, homologies with the human protein as bits per aminoacid (bpa). On the x axis, approximate time of appearance in million of years.
The graphic shows the big information jump in vertebrates for all three protens , especially CARD11.
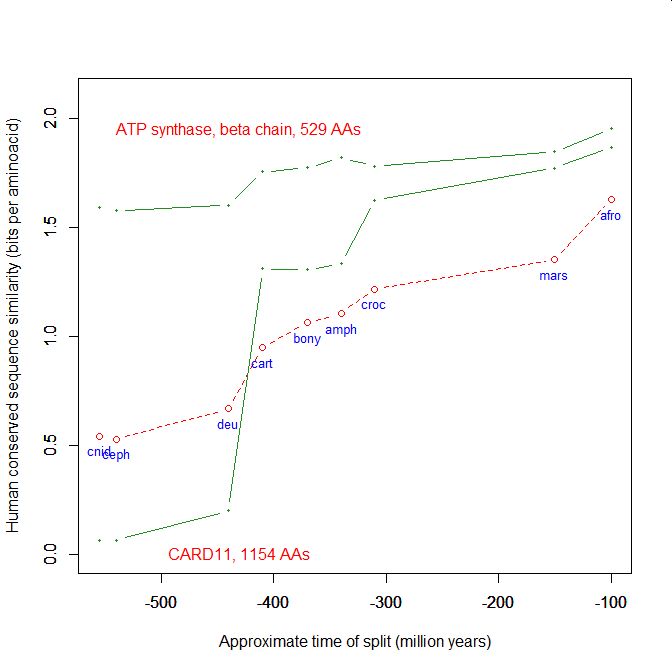
Added graphic: two very different proteins and their functional history