A week ago, VJT put up a useful set of excerpts from Axe’s 2010 paper on proteins and barriers they pose to Darwinian, blind watchmaker thesis evolution. During onward discussions, it proved useful to focus on some excerpts where Axe spoke to some numerical considerations and the linked idea of islands of specific function deeply isolated in AA sequence and protein fold domain space, though he did not use those exact terms.
I think it worth the while to headline the clips, for reference (instead of leaving them deep in a discussion thread):
_________________
ABSTRACT: >> Four decades ago, several scientists suggested that the impossibility of any evolutionary process sampling anything but a miniscule fraction of the possible protein sequences posed a problem for the evolution of new proteins. This potential problem—the sampling problem—was largely ignored, in part because those who raised it had to rely on guesswork to fill some key gaps in their understanding of proteins. The huge advances since that time call for a care -ful reassessment of the issue they raised. Focusing specifically on the origin of new protein folds, I argue here that the sampling problem remains. The difficulty stems from the fact that new protein functions, when analyzed at the level of new beneficial phenotypes, typically require multiple new protein folds, which in turn require long stretches of new protein sequence. Two conceivable ways for this not to pose an insurmountable barrier to Darwinian searches exist. One is that protein function might generally be largely indifferent to protein sequence. The other is that rela-tively simple manipulations of existing genes, such as shuffling of genetic modules, might be able to produce the necessary new folds. I argue that these ideas now stand at odds both with known principles of protein structure and with direct experimental evidence . . . >>
Pp 5 – 6: >> . . . we need to quantify a boundary value for m, meaning a value which, if exceeded, would solve the whole sampling problem. To get this we begin by estimating the maximum number of opportunities for spontane-ous mutations to produce any new species-wide trait, meaning a trait that is fixed within the population through natural selection (i.e., selective sweep). Bacterial species are most conducive to this because of their large effective population sizes. 3 So let us assume, generously, that an ancient bacterial species sustained an effective population size of 10 ^10 individuals [26] while passing through 10^4 generations per year. After five billion years, such a species would produce a total of 5 × 10 ^ 23 (= 5 × 10^ 9 x 10^4 x 10 ^10 ) cells that happen (by chance) to avoid the small-scale extinction events that kill most cells irrespective of fitness. These 5 × 10 ^23 ‘lucky survivors’ are the cells available for spontaneous muta-tions to accomplish whatever will be accomplished in the species. This number, then, sets the maximum probabilistic resources that can be expended on a single adaptive step. Or, to put this another way, any adaptive step that is unlikely to appear spontaneously in that number of cells is unlikely to have evolved in the entire history of the species.
In real bacterial populations, spontaneous mutations occur in only a small fraction of the lucky survivors (roughly one in 300 [27]). As a generous upper limit, we will assume that all lucky survivors happen to receive mutations in portions of the genome that are not constrained by existing functions 4 , making them free to evolve new ones. At most, then, the number of different viable genotypes that could appear within the lucky survivors is equal to their number, which is 5 × 10^ 23 . And again, since many of the genotype differences would not cause distinctly new proteins to be produced, this serves as an upper bound on the number of new protein sequences that a bacterial species may have sampled in search of an adaptive new protein structure.
Let us suppose for a moment, then, that protein sequences that produce new functions by means of new folds are common enough for success to be likely within that number of sampled sequences. Taking a new 300-residue structure as a basis for calculation (I show this to be modest below), we are effectively supposing that the multiplicity factor m introduced in the previous section can be as large as 20 ^300 / 5×10^ 23 ~ 10 ^366 . In other words, we are supposing that particular functions requiring a 300-residue structure are real-izable through something like 10 ^366 distinct amino acid sequences. If that were so, what degree of sequence degeneracy would be implied? More specifically, if 1 in 5×10 23 full-length sequences are supposed capable of performing the function in question, then what proportion of the twenty amino acids would have to be suit-able on average at any given position? The answer is calculated as the 300 th root of (5×10 23 ) -1 , which amounts to about 83%, or 17 of the 20 amino acids. That is, by the current assumption proteins would have to provide the function in question by merely avoid-ing three or so unacceptable amino acids at each position along their lengths.
No study of real protein functions suggests anything like this degree of indifference to sequence. In evaluating this, keep in mind that the indifference referred to here would have to charac-terize the whole protein rather than a small fraction of it. Natural proteins commonly tolerate some sequence change without com- plete loss of function, with some sites showing more substitutional freedom than others. But this does not imply that most mutations are harmless. Rather, it merely implies that complete inactivation with a single amino acid substitution is atypical when the start-ing point is a highly functional wild-type sequence (e.g., 5% of single substitutions were completely inactivating in one study [28]). This is readily explained by the capacity of well-formed structures to sustain moderate damage without complete loss of function (a phenomenon that has been termed the buffering effect [25]). Conditional tolerance of that kind does not extend to whole proteins, though, for the simple reason that there are strict limits to the amount of damage that can be sustained.
A study of the cumulative effects of conservative amino acid substitutions, where the replaced amino acids are chemically simi-lar to their replacements, has demonstrated this [23]. Two unrelat-ed bacterial enzymes, a ribonuclease and a beta-lactamase, were both found to suffer complete loss of function in vivo at or near the point of 10% substitution, despite the conservative nature of the changes. Since most substitutions would be more disruptive than these conservative ones, it is clear that these protein functions place much more stringent demands on amino acid sequences than the above supposition requires.
Two experimental studies provide reliable data for estimating the proportion of protein sequences that perform specified func -tions [–> note the terms] . One study focused on the AroQ-type chorismate mutase, which is formed by the symmetrical association of two identical 93-residue chains [24]. These relatively small chains form a very simple folded structure (Figure 5A). The other study examined a 153-residue section of a 263-residue beta-lactamase [25]. That section forms a compact structural component known as a domain within the folded structure of the whole beta-lactamase (Figure 5B). Compared to the chorismate mutase, this beta-lactamase do-main has both larger size and a more complex fold structure.
In both studies, large sets of extensively mutated genes were produced and tested. By placing suitable restrictions on the al-lowed mutations and counting the proportion of working genes that result, it was possible to estimate the expected prevalence of working sequences for the hypothetical case where those restric-tions are lifted. In that way, prevalence values far too low to be measured directly were estimated with reasonable confidence.
The results allow the average fraction of sampled amino acid substitutions that are functionally acceptable at a single amino acid position to be calculated. By raising this fraction to the power l, it is possible to estimate the overall fraction of working se-quences expected when l positions are simultaneously substituted (see reference 25 for details). Applying this approach to the data from the chorismate mutase and the beta-lactamase experiments gives a range of values (bracketed by the two cases) for the preva-lence of protein sequences that perform a specified function. The reported range [25] is one in 10 ^77 (based on data from the more complex beta-lactamase fold; l = 153) to one in 10 ^53 (based on the data from the simpler chorismate mutase fold, adjusted to the same length: l = 153). As remarkable as these figures are, par-ticularly when interpreted as probabilities, they were not without precedent when reported [21, 22]. Rather, they strengthened an existing case for thinking that even very simple protein folds can place very severe constraints on sequence. [–> Islands of function issue.]
Rescaling the figures to reflect a more typical chain length of 300 residues gives a prevalence range of one in 10 ^151 to one in 10 ^104 . On the one hand, this range confirms the very highly many-to-one mapping of sequences to functions. The corresponding range of m values is 10 ^239 (=20 ^300 /10 ^151 ) to 10 ^286 (=20 ^300 /10 ^104 ), meaning that vast numbers of viable sequence possibilities exist for each protein function. But on the other hand it appears that these functional sequences are nowhere near as common as they would have to be in order for the sampling problem to be dis-missed. The shortfall is itself a staggering figure—some 80 to 127 orders of magnitude (comparing the above prevalence range to the cutoff value of 1 in 5×10 23 ). So it appears that even when m is taken into account, protein sequences that perform particular functions are far too rare to be found by random sampling.>>
Pp 9 – 11: >> . . . If aligned but non-matching residues are part-for-part equivalents, then we should be able to substitute freely among these equivalent pairs without impair-ment. Yet when protein sequences were even partially scrambled in this way, such that the hybrids were about 90% identical to one of the parents, none of them had detectable function. Considering the sensitivity of the functional test, this implies the hybrids had less than 0.1% of normal activity [23]. So part-for-part equiva-lence is not borne out at the level of amino acid side chains.
In view of the dominant role of side chains in forming the bind-ing interfaces for higher levels of structure, it is hard to see how those levels can fare any better. Recognizing the non-generic [–> that is specific and context sensitive] na-ture of side chain interactions, Voigt and co-workers developed an algorithm that identifies portions of a protein structure that are most nearly self-contained in the sense of having the fewest side-chain contacts with the rest of the fold [49]. Using that algorithm, Meyer and co-workers constructed and tested 553 chimeric pro-teins that borrow carefully chosen blocks of sequence (putative modules) from any of three natural beta lactamases [50]. They found numerous functional chimeras within this set, which clearly supports their assumption that modules have to have few side chain contacts with exterior structure if they are to be transport-Able.
At the same time, though, their results underscore the limita-tions of structural modularity. Most plainly, the kind of modular-ity they demonstrated is not the robust kind that would be needed to explain new protein folds. The relatively high sequence simi-larity (34–42% identity [50]) and very high structural similarity of the parent proteins (Figure 8) favors successful shuffling of modules by conserving much of the overall structural context. Such conservative transfer of modules does not establish the ro-bust transportability that would be needed to make new folds. Rather, in view of the favorable circumstances, it is striking how low the success rate was. After careful identification of splice sites that optimize modularity, four out of five tested chimeras were found to be completely non-functional, with only one in nine being comparable in activity to the parent enzymes [50]. In other words, module-like transportability is unreliable even under extraordinarily favorable circumstances [–> these are not generally speaking standard bricks that will freely fit together in any freely plug- in compatible pattern to assemble a new structure] . . . .
Graziano and co-workers have tested robust modularity directly by using amino acid sequences from natural alpha helices, beta strands, and loops (which connect helices and/or strands) to con-struct a large library of gene segments that provide these basic structural elements in their natural genetic contexts [52]. For those elements to work as robust modules, their structures would have to be effectively context-independent, allowing them to be com-bined in any number of ways to form new folds. A vast number of combinations was made by random ligation of the gene segments, but a search through 10^8 variants for properties that may be in-dicative of folded structure ultimately failed to identify any folded proteins. After a definitive demonstration that the most promising candidates were not properly folded, the authors concluded that “the selected clones should therefore not be viewed as ‘native-like’ proteins but rather ‘molten-globule-like’” [52], by which they mean that secondary structure is present only transiently, flickering in and out of existence along a compact but mobile chain. This contrasts with native-like structure, where secondary structure is locked-in to form a well defined and stable tertiary Fold . . . .
With no discernable shortcut to new protein folds, we conclude that the sampling problem really is a problem for evolutionary accounts of their origins. The final thing to consider is how per-vasive this problem is . . . Continuing to use protein domains as the basis of analysis, we find that domains tend to be about half the size of complete protein chains (compare Figure 10 to Figure 1), implying that two domains per protein chain is roughly typical. This of course means that the space of se-quence possibilities for an average domain, while vast, is nowhere near as vast as the space for an average chain. But as discussed above, the relevant sequence space for evolutionary searches is determined by the combined length of all the new domains needed to produce a new beneficial phenotype. [–> Recall, courtesy Wiki, phenotype: “the composite of an organism’s observable characteristics or traits, such as its morphology, development, biochemical or physiological properties, phenology, behavior, and products of behavior (such as a bird’s nest). A phenotype results from the expression of an organism’s genes as well as the influence of environmental factors and the interactions between the two.”]
As a rough way of gauging how many new domains are typi-cally required for new adaptive phenotypes, the SUPERFAMILY database [54] can be used to estimate the number of different protein domains employed in individual bacterial species, and the EcoCyc database [10] can be used to estimate the number of metabolic processes served by these domains. Based on analysis of the genomes of 447 bacterial species 11, the projected number of different domain structures per species averages 991 (12) . Compar-ing this to the number of pathways by which metabolic processes are carried out, which is around 263 for E. coli,13 provides a rough figure of three or four new domain folds being needed, on aver-age, for every new metabolic pathway 14 . In order to accomplish this successfully, an evolutionary search would need to be capable of locating sequences that amount to anything from one in 10 ^159 to one in 10 ^308 possibilities 15 , something the neo-Darwinian model falls short of by a very wide margin. >>
____________________
Those who argue for incrementalism or exaptation and fortuitous coupling or Lego brick-like modularity or the like need to address these and similar issues. END
PS: Just for the objectors eager to queue up, just remember, the Darwinism support essay challenge on actual evidence for the tree of life from the root up to the branches and twigs is still open after over two years, with the following revealing Smithsonian Institution diagram showing the first reason why, right at the root of the tree of life:
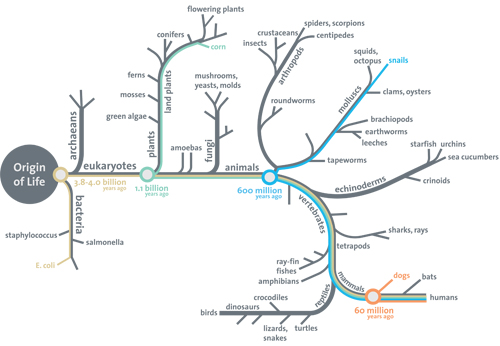
No root, no shoots, folks. (Where, the root must include a viable explanation of gated encapsulation, protein based metabolism and cell functions, code based protein assembly and the von Neumann self replication facility keyed to reproducing the cell.)