In recent posts, I have been discussing some important points about the reasonable meaning of homologies and differences in the proteome in the course of natural history. For the following discussion, just to be clear, I will accept a scenario of Common Descent (as explained in many recent posts) in the context of an ID approach. I will also accept the very reasonable concept that neutral or quasi-neutral random variation happens in time, and that negative (purifying) selection is the main principle which limits random variation in functional sequences.
My main points are the following:
- Given those premises, homologies through natural history are certainly an indicator of functional constraints, because they mean that some sequence cannot be significantly transformed by random variation. Another way to express this concept is that variation in a functional sequence with strong functional constraints is not neutral, but negative, and therefore negative selection will in mot cases suppress variation and conserve the functional sequence through time. This is a very important point, because it means that strong homologies through time point to high functional complexity, and therefore to design. I have used this kind of argument, for example, for proteins like the beta chain of ATP synthase (highly conserved from LUCA to humans) and Histone H3 (highly conserved in all eukaryotes).
- Differences between homologues, instead, can have two completely different meanings:
- 2a) They can be the result of accumulating neutral variation in parts of the molecule which are not functionally constrained
- 2b) They can be the expression of differences in function in different species and contexts
I do believe that both 2a and 2b happen and have an important role in shaping the proteome. 2b, in particular, is often underestimated. It is also, in many cases, a very good argument for ID.
Now, I will try to apply this reasoning to one example. I have chosen a regulatory protein, one which is not really well understood, but which has certainly an important role in epigenetic regulation. The protein is called “Prickle”, and we will consider in particular the one known as “Prickle 1”. It has come to my attention trough an interesting paper linked by Dionisio (to whom go my sincere thanks and appreciation):
In brief, Prickle is a molecule implied, among other things, in planar polarization events and in the regulation of neural system in vertebrates.
Let’s have a look at the protein. From Wikipedia:
Prickle is part of the non-canonical Wnt signaling pathway that establishes planar cell polarity.[2] A gain or loss of function of Prickle1 causes defects in the convergent extension movements of gastrulation.[3] In epithelial cells, Prickle2 establishes and maintains cell apical/basal polarity.[4] Prickle1 plays an important role in the development of the nervous system by regulating the movement of nerve cells.[5
And:
Mutations in Prickle genes can cause epilepsy in humans by perturbing Prickle function.[12] One mutation in Prickle1 gene can result in Prickle1-Related Progressive Myoclonus Epilepsy-Ataxia Syndrome.[2] This mutation disrupts the interaction between prickle-like 1 and REST, which results in the inability to suppress REST.[2] Gene knockdown of Prickle1 by shRNA or dominant-negative constructs results in decreased axonal and dendritic extension in neurons in the hippocampus.[5] Prickle1 gene knockdown in neonatal retina causes defects in axon terminals of photoreceptors and in inner and outer segments.[5]
The human protein is 831 AAs long.
Its structure is interesting: according to Uniprot, in the first part of the molecule we can recognize 4 domains:
1 PET domain: AAs 14 – 122
3 LIM zinc-binding doamins: AAs 124 – 313
In the rest of the sequence (AAs 314 – 831) no known domain is recognized.
Here is the FASTA sequence of the human protein, divided in the two parts (red: 4 domain part; blue: no domain part):
>sp|Q96MT3|PRIC1_HUMAN Prickle-like protein 1 OS=Homo sapiens GN=PRICKLE1 PE=1 SV=2 MPLEMEPKMSKLAFGCQRSSTSDDDSGCALEEYAWVPPGLRPEQIQLYFACLPEEKVPYV NSPGEKHRIKQLLYQLPPHDNEVRYCQSLSEEEKKELQVFSAQRKKEALGRGTIKLLSRA VMHAVCEQCGLKINGGEVAVFASRAGPGVCWHPSCFVCFTCNELLVDLIYFYQDGKIHCG RHHAELLKPRCSACDEIIFADECTEAEGRHWHMKHFCCLECETVLGGQRYIMKDGRPFCC GCFESLYAEYCETCGEHIGVDHAQMTYDGQHWHATEACFSCAQCKASLLGCPFLPKQGQI YCSKTCSLGEDVHASDSSDSAFQSARSRDSRRSVRMGKSSRSADQCRQSLLLSPALNYKF PGLSGNADDTLSRKLDDLSLSRQGTSFASEEFWKGRVEQETPEDPEEWADHEDYMTQLLL KFGDKSLFQPQPNEMDIRASEHWISDNMVKSKTELKQNNQSLASKKYQSDMYWAQSQDGL GDSAYGSHPGPASSRRLQELELDHGASGYNHDETQWYEDSLECLSDLKPEQSVRDSMDSL ALSNITGASVDGENKPRPSLYSLQNFEEMETEDCEKMSNMGTLNSSMLHRSAESLKSLSS ELCPEKILPEEKPVHLPVLRRSKSQSRPQQVKFSDDVIDNGNYDIEIRQPPMSERTRRRV YNFEERGSRSHHHRRRRSRKSRSDNALNLVTERKYSPKDRLRLYTPDNYEKFIQNKSARE IQAYIQNADLYGQYAHATSDYGLQNPGMNRFLGLYGEDDDSWCSSSSSSSDSEEEGYFLG QPIPQPRPQRFAYYTDDLSSPPSALPTPQFGQRTTKSKKKKGHKGKNCIIS
So, this is a very interesting situation, which is not so rare. We have the first part of the sequence (313 AAs) which configures well known and conserved domains, while “the rest”(517 AAs) is apparently not understood in terms of structure and function.
So, to better understand what all this could mean, I have blasted those two parts of the human molecule separately.
(Those who are not interested in the technical details, can choose here to go on to the conclusions 🙂 )
The first part of the sequence (AAs 1 – 313) shows no homologies in prokaryotes. So, we are apparently in the presence of domains which appear in eukaryotes.
In fungi, we find some significant, but weak, homologues. The best hit is an expect of 2e-21, with 56 identities and 93 positives (99.4 bits).
Multicellular organisms have definitely stronger homologies:
C. elegans: 144 identities, 186 positives, expect 2e-90 (282 bits)
Drosophila melanogaster: 202 identities, 244 positives, expect 5e-152 (447 bits)
Let’s go to non vertebrate chordates:
Cephalochordata (Branchiostoma floridae): 222 identities, 256 positives, expect 6e-165 (484 bits)
Tunicata (Ciona intestinalis): 196 identities, 241 positives, expect 2e-149 (442 bits)
Now, vertebrates:
Cartilaginous fishes (Callorhincus milii): 266 identities, 290 positives, expect 0.0 (588 bits)
Bony fishes (Lepisosteus oculatus): 274 identities, 292 positives, expect 0.0 (598 bits)
Mammals (Mouse): 309 identities, 312 positives, expect 0.0 (664 bits)
IOWs, what we see here is that the 4 domain part of the molecule, absent in prokaryotes, is already partially observable in single celled eukaryotes, and is strongly recognizable in all multicellular beings. It is interesting that homology with the human form is not very different between drosophila and non vertebrate chordates, while there is a significant increase in vertebrates, and practical identity already in mouse. That is a very common pattern, and IMO it can be explained as a mixed result of different functional constraints and neutral evolution in different time splits.
Now, let’s go to “the rest” of the molecule: AAs 314 – 831 (518 AAs). No recognizable domains here.
What is the behaviour of this sequence in natural history?
Again, let’s start again from the human sequence and blast it.
With Prokaryotes: no homologies
With Fungi: no homologies
C. elegans: no homologies
Drosophila melanogaster: no homologies
Let’s go to non vertebrate chordates:
Cephalochordata (Branchiostoma floridae): no significant homologies
Tunicata (Ciona intestinalis): no significant homologies
So, there is no significant homology in the whole range of eukaryotes, excluding vertebrates and including chordates which are not vertebrates.
Now, what happens with vertebrates?
Here are the numbers:
Cartilaginous fishes (Callorhincus milii): 350 identities, 429 positives, expect 0.0 (597 bits)
Bony fishes (Lepisosteus oculatus): 396 identities, 446 positives, expect 0.0 (662 bits)
Mammals (Mouse): 466 identities, 491 positives, expect 0.0 (832 bits)
IOWs, what we see here is that the no domain part of the molecule is practically non existent in prokaryotes, in single celled eukaryotes and in all multicellular beings which are not vertebrates. In vertebrates, the sequence is not only present in practically all vertebrates, but it is also extremely conserved, from sharks to humans. So, we have a steep informational jump from non chordates and non vertebrate chordates, where the sequence is practically absent, to the very first vertebrates, where the sequence is already highly specific.
What does that mean from an ID point of view? It’s simple:
a) The sequence of 517 AAs which represents the major part of the human protein must be reasonably considered highly functional, because it is strongly conserved throughout vertebrate evolution. As we have said in the beginning, the only reasonable explanation for high conservation throughout a span of time which must be more than 400 million years long is the presence of strong functional constraints in the sequence.
b) The sequence and its function, whatever it may be (but it is probably an important regulatory function) is highly specific of vertebrates.
We have here a very good example of a part of a protein which practically appears in vertebrates while it is absent before, and which is reasonably highly functional in vertebrates.
So, to sum up:
- Prickle 1 is a functional protein which is found in all eukaryotes.
- The human sequence can be divided in two parts, with different properties.
- The first part, while undergoing evolutionary changes, is rather well conserved in all eukaryotes. Its function can be better understood, because it is made of known domains with known structure.
- The second part does not include any known domain or structure, and is practically absent in all eukaryotes except vertebrates.
- In vertebrates, it is highly conserved and almost certainly highly functional. Probably as a regulatory epigenetic sequence.
- For its properties, this second part, and its functional sequence, are a very reasonable object for a strong design inference.
I have added a graph to show better what is described in the conclusions, in particular the information jump in vertebrates for the second part of the sequence:
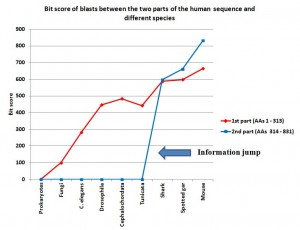
Note: Thanks to the careful checking of Alicia Cartelli, I have corrected a couple of minor imprecisions in the data and the graph (see posts #83 and #136). Thank you, Alicia, for your commitment. The sense of the post, however, does not change.
Those who are interested in the evolutionary behaviour of protein Prickle 2 could give a look at my posts #127 and #137.