BA77 often posts clips of citations and links here at UD. After a recent noticeable break (we missed you), he has just [–> correction: he posted in a thread some time ago which just got a comment from TJG . . . ] posted a link to a video on objections to prof Dawkins’ claims that FOXP 2 (let me be exact) etc trees give the same structure:
[youtube IfFZ8lCn5uU]
Key clips include a transcript:
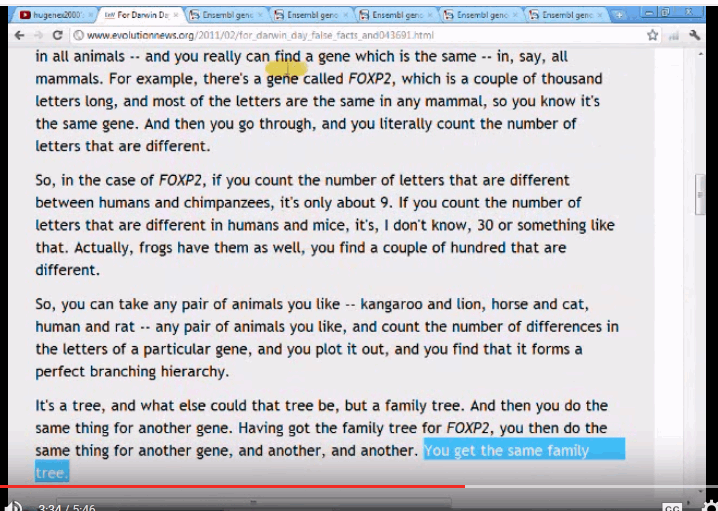
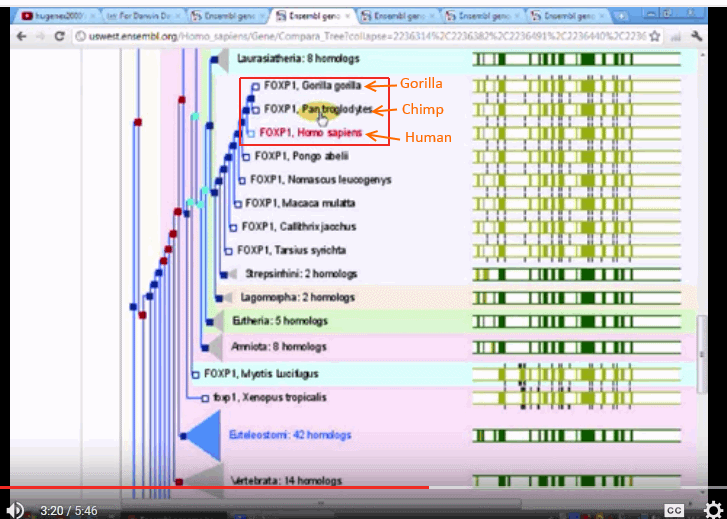
Plus, several family trees, such as FOXP1, showing:
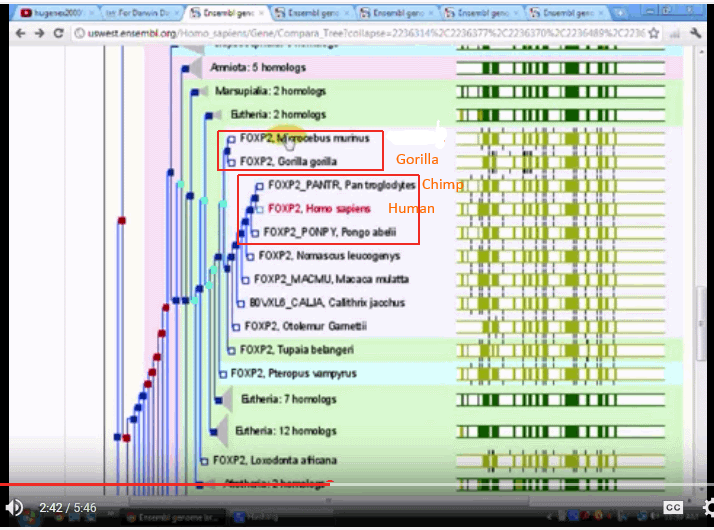
With FOXP2:
 FOXP3:
FOXP3:
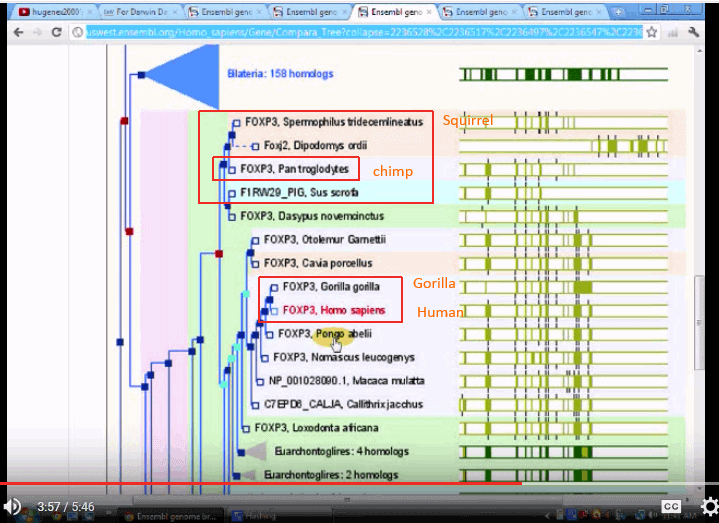
The three trees seem to be quite divergent, one putting chimps with squirrels and the like, another putting gorillas on a different branch, and only one putting the three on neighbouring twigs.
This seems to be consistent with the objection that molecular trees are both inconsistent with traditional trees and that they are mutually inconsistent.
So, has Mr Dawkins erred, or is this the grand-daddy of all elaborate Creationist quote mines and hoaxes?
If you assert or imply the latter, what is your evidence? END