With this OP, I am starting a series (I hope) of articles whose purpose is to present interesting proteins which can be of specific relevance to ID theory, for their functional context and evolutionary history.
DNA-binding protein SATB1
SATB1 (accession number Q01826) is a very intriguing molecule. Let’s start with some information we can find at Uniprot, a fundamental protein database, about what is known of its function (in the human form):
Crucial silencing factor contributing to the initiation of X inactivation mediated by Xist RNA that occurs during embryogenesis and in lymphoma
And:
Transcriptional repressor controlling nuclear and viral gene expression in a phosphorylated and acetylated status-dependent manner, by binding to matrix attachment regions (MARs) of DNA and inducing a local chromatin-loop remodeling. Acts as a docking site for several chromatin remodeling enzymes
IOWs, it is an important regulatory protein involved in many different, and not necessarily well understood, processes, which binds to DNA and in involved in chromatin remodeling.
It is also involved in hematopoiesis (especially in T cell development), and has important roles in the biology of some tumors:
Modulates genes that are essential in the maturation of the immune T-cell CD8SP from thymocytes. Required for the switching of fetal globin species, and beta- and gamma-globin genes regulation during erythroid differentiation. Plays a role in chromatin organization and nuclear architecture during apoptosis.
Reprograms chromatin organization and the transcription profiles of breast tumors to promote growth and metastasis.
Keywords for molecular function: Chromatin regulator, DNA-binding, Repressor
Now, some information about the protein itself. I will relate, again, to the human form of the protein:
Length: 763 AAs. It’s a rather big protein, like many important regulatory molecules.
Its subcellular location is in the nucleus.
It is a multi-domain protein, with at least 5 detectable domains and many DNA binding sites.
Evolutionary history of SATB1
Now, let’s see some features of the evolutionary history of this protein in the course of metazoa evolution.
I will use here the same tools that I have developed and presented in my previous OP:
The amazing level of engineering in the transition to the vertebrate proteome: a global analysis
So, I invite all those who are interested in the technical details to refer to that OP.
Here is a graph of the levels of homology to the human protein detectable in other metazoan groups, expressed as mean bitscore per aminoacid site:
Fig. 1: Evolutionary history of SATB1 by human-conserved functional information
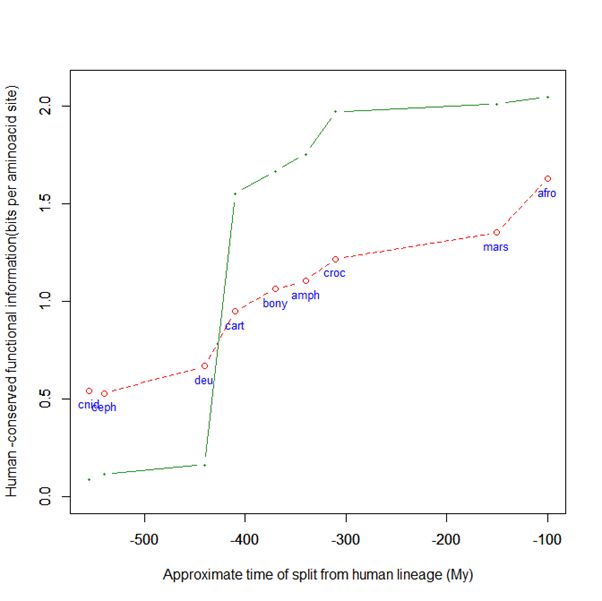
The green line represents the evolutionary history of our protein, while the red dotted line is the reference mean line for the groups considered, as already presented in my previous post quoted above (Fig. 2).
As everyone can see, this specific protein has a very sudden gain in human-conserved information with the transition from pre-vertebrates to vertebrates. So, it represents a very good example of the information jump that I have tried to quantify globally in my previous post.
Here, the jump is of almost 1.5 bits per aminoacid site. What does that mean?
Let’s remember that the protein is 763 AA long. Therefore, an increase of information of 1.5 bits per aminoacid corresponds to more than 1000 bits of information. To be precise, the jump from the best pre-vertebrate hit to the best hit in cartilaginous fish is:
1049 bits
But let’s see more in detail how the jump happens.
I will show here in detail some results of protein blasts. All of them have been obtained using the Blastp software at the NCBI site:
https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastp&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome
with default settings.
Here is the result of blasting the human protein against all known protein sequences except for vertebrate sequences:
Fig. 2: Results of blasting human SATB1 against all non vertebrate protein sequences
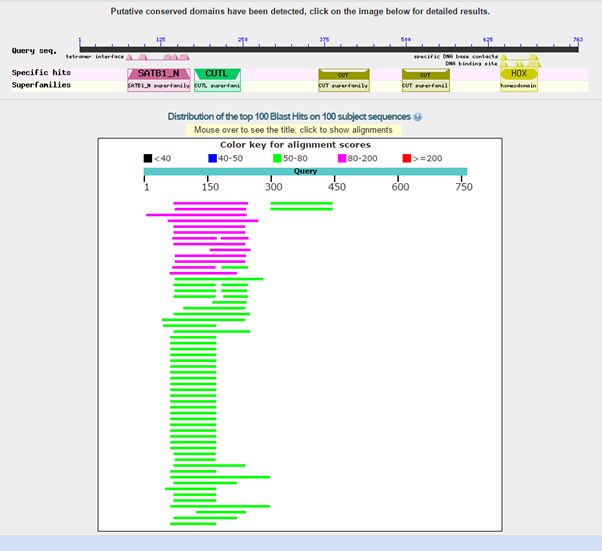
As can be seen, we find only low homologies in non vertebrates, and they are essentially restricted to a small part of the molecule, that correspond to the first two domains in the protein, or just to the first domain. The image shows clearly that all the rest of the sequence has no detectable significant homologies in non vertebrates (except for a couple of very low homologies for the third domain).
The best hit in non vertebrates is 154 bits with Parasteatoda tepidariorum, a spider. Here it is:
Fig. 3: The best hit in non vertebrates (with a spider)
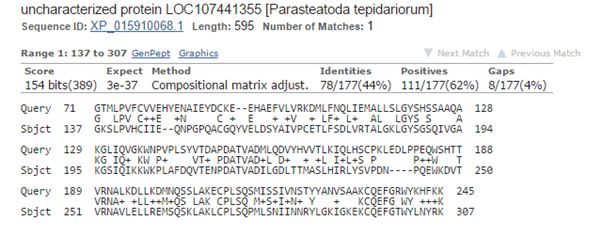
The upper line (Query) is the human sequence. The bottom line (Sbjct) is the aligned sequence of the spider. In the middle line, letters are identities, “+” characters are similarities (substitutions which are frequently observed in proteins, and are probably quasi-neutral), and empty spaces are less frequent substitutions, those that are more likely to affect protein structure and function if they happen at a functionally important aminoacid site.
The alignment here is absolutely restricted to AAs 71 – 245 (the first two domains), and involves only 177 AAs. Of these, only 78 (44%) are identities and 111 (62%) are positives (identities + similarities). So, in the whole protein we have only 78 identities out of 763 (10.2%).
The spider protein is labeled as “uncharacterized protein”, and that is the case in most of the other non vertebrate hits.
All the other non vertebrate hits, with a couple of exceptions, are well below 100 bits, most of them between 70 and 86 bits.
IOWs, the protein as we know it in vertebrates essentially does not exist in non vertebrates.
Even non vertebrate deuterostomia, which should be the nearest precursors of the first vertebrates, have extremely low homology bitscores with the human protein:
Saccoglossus kowalevskii (hemichordates): 87 bits
Branchiostoma floridae (cephalochordate): 67 bits
The information jump in vertebrates
Now, what happens with the first vertebrates?
The oldest split in vertebrates is the one between cartilaginous fish and bony fish (from which the human lineage derives). Therefore, homologies that are conserved between cartilaginous fish and humans had reasonably to be already present in the Last Common Ancestor of Vertebrates, before the split between cartilaginous fish and bony fish, and have been conserved for about 420 million years.
So, let’s see the best hit between the human protein and cartilaginous fish. It is with Rhincodon typus (whale shark). Here it is:
Fig. 4: The best hit of human SATB1 in cartilaginous fish (with the whale shark)

Here, the alignment involves practically the whole molecule (756 AAs), and we have 1203 bits of homology, 603 identities (79%), 659 positives (86%).
IOWs, the two molecules are almost identical. And the homology is extremely high not only in the domain parts, but also in the rest of the protein sequence.
Now, the evolutionary time between pre-vertebrates and the first split in vertebrates is certainly rather small, a few million years, or at most 20 – 30 million years. Not a big chronological window at all, in evolutionary terms.
However, in that window, this protein appears almost complete. 603 aminoacids are already those that will remain up to the human form of the protein, and only 78 of them were detectable in the best hit before vertebrate appearance.
1049 bits of new, original functional information. In such a short evolutionary window.
Functionality
Why functional? Because those 603 aminoacid have remained the same thorugh more than 400 million years of evolution. They have evaded neutral or quasi neutral variation, that would have certainly completely transformed the sequence in such a big evolutionary time, if those aminoacid sites were not under extreme functional constraint and purifying (negative) selection.
Now, I say that this fact cannot in any way be explained by any neo-darwinian model. Absolutely not.
Moreover, there is absolutely no evidence in the available proteome of any intermediate form, of any gradual development of the functional sequence that will be conserved up to humans (except, of course, for the 50 – 78 AAs which are already detectable in the first two domains in many pre -vertebrates).
By the way, Callorhincus milii, the Elephant shark, has almost identical values of homology:
1184 bits, 599 identities, 654 positives
But, how important is this protein?
In the ExAC database, a database of variations in the human genome, missense mutations are 110 out of 260.3 expected, with a z score of 4.56, an extremely high measure of functional constraint.
The recent medical literature has a lot of articles about the important role of SATB1 at least in two big fields:
- T cell development
- Tumor development (many different kinds of tumors)
If we want to sum up in a few words what is known, we could say that SATB1 is considered a master regulator, essentially a complex transcription repressor, involved mainly (but not only) in the development of the immune system, in particular T cells. A disregulation of this protein is linked to many aspects of tumor invasivity (especially metastases). The protein seems to act, among other possibilities, as a global organizer of chromatin states.
Here is a very brief recent bibliography:
Essential Roles of SATB1 in Specifying T Lymphocyte Subsets
SATB1-mediated Functional Packaging of Chromatin into Loops
DNA-binding protein SATB2
But there is more. There is another protein which is very similar to SATB1. It is called DNA-binding protein SATB2 (accession number Q9UPW6).
Its length is very similar to SATB1: 733 AAs.
Uniprot describes its function as follows:
Binds to DNA, at nuclear matrix- or scaffold-associated regions. Thought to recognize the sugar-phosphate structure of double-stranded DNA. Transcription factor controlling nuclear gene expression, by binding to matrix attachment regions (MARs) of DNA and inducing a local chromatin-loop remodeling. Acts as a docking site for several chromatin remodeling enzymes
Which is very similar to SATB1. But now come the differences. While SATB1 is implied prevalently in T cell development and tumor development, SATB2 is:
Required for the initiation of the upper-layer neurons (UL1) specific genetic program and for the inactivation of deep-layer neurons (DL) and UL2 specific genes, probably by modulating BCL11B expression. Repressor of Ctip2 and regulatory determinant of corticocortical connections in the developing cerebral cortex. May play an important role in palate formation. Acts as a molecular node in a transcriptional network regulating skeletal development and osteoblast differentiation
So, similar proteins with rather different specificities. While SATB1 is mainly connexted to adaptive immunity (T cell development), SATB2 seems to be more linked to neuronal development. Like SATB1, it is involved in cancer development, although usually in different types of cancer.
Here is a brief recent bibliography about SATB2:
SATB1 and SATB2 play opposing roles in c-Myc expression and progression of colorectal cancer
However, how similar is SATB2 to SATB1 in terms of sequence homology?
Here is a direct blast of the two human molecules:
Fig. 5: Blast of human SATB1 vs human SATB2:

OK, they are very similar, but… only 460 identities, 550 positives, 854 bits. IOWs, these two human proteins are similar, but not so similar as the two sequences of SATB1 in the shark and in humans.
Now, here is the evolutionary history of SATB2:
Fig. 6: Evolutionary history of SATB2 by human-conserved functional information
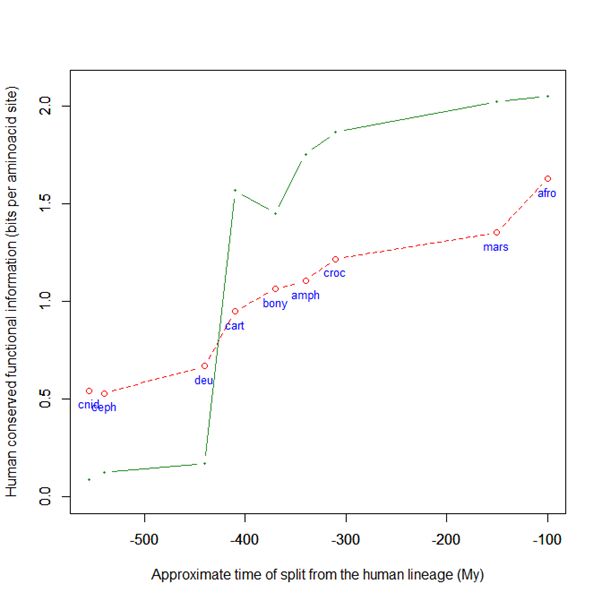
As everyone can see, it is almost identical to the evolutionary history of SATB1. To see it even better, Fig. 7 shows the two evolutionary histories together (the green line is SATB1, the brown line is SATB2):
Fig. 7: Evolutionary history of SATB1 and SATB2 by human-conserved functional information
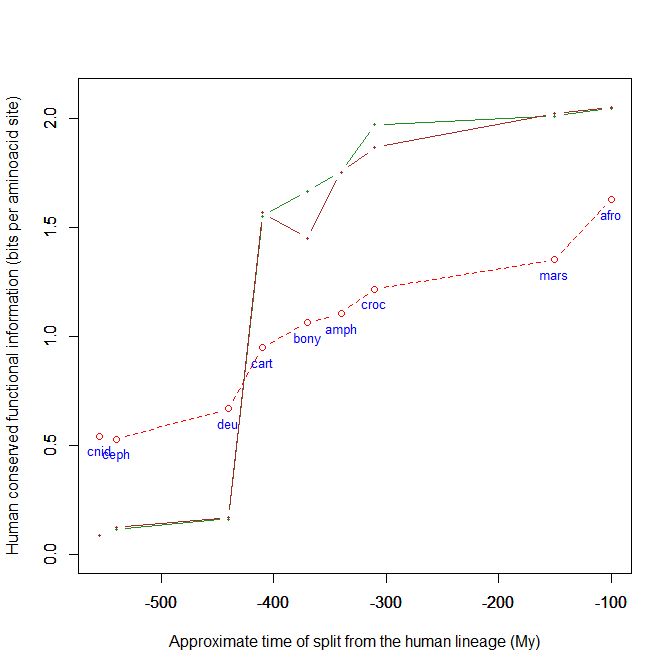
In particular, pre-vertebrate history and the jump in cartilaginous fish are practically identical. And yet these are two different molecules, as we have seen, with different specificities and about one third of difference in sequence.
Now, let’s blast human SATB2 against cartilaginous fish. Again the best hit is with the whale shark:
Fig. 8: The best hit of human SATB2 in cartilaginous fish (with the whale shark)
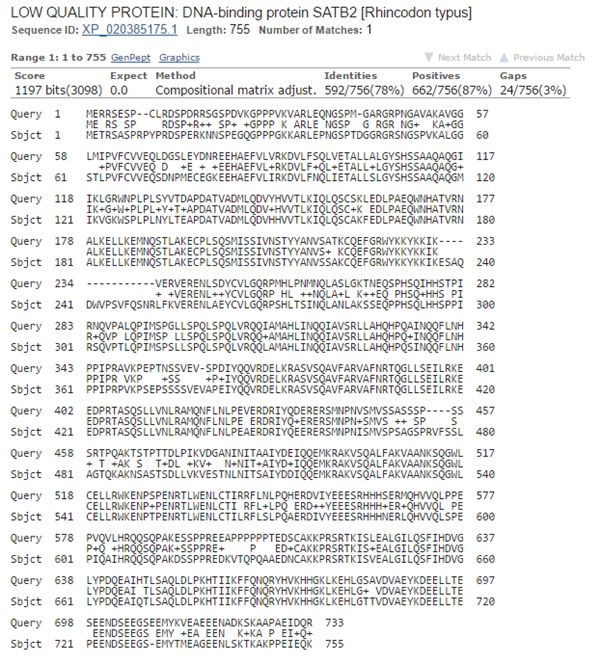
And the numbers are very similar, incredibly similar I would say, to those we found for SATB1:
1197 bits, 592 identities, 662 positives.
But what if we blast SATB1 of the whale shark against SATB2 of the whale shark?
Here are the results:
Fig. 9: Blast of whale shark SATB1 vs whale shark SATB2:

Now, please, compare the numbers we got here with those from the similar blast between the two proteins in humans:
SATB1 human vs SATB2 human: 460 identities, 550 positives, 854 bits
SATB1 shark vs SATB2 shark: 468 identities, 556 positives, 856 bits
Almost exactly the same numbers! Wow!
What does that mean?
It means that this system of two similar proteins with different function arises in vertebrates as a whole system, already complete, with the two components already differentiated, and is conserved almost identical up to humans. Indeed, SATB1 and SATB2 have the same degree of homology both in sharks and in humans, and the two SATB1 proteins in shark and humans, as well as the two SATB2 proteins in shark and humans, have greater similarity, after more than 400 million years of divergence, than SATB1 and SATB2 show when compared, both in sharks and in humans.
Would you describe that as sudden appearance of huge amounts of functional information, followed by an extremely long stasis? I certainly would!
The following table sums up these results:
| Sequence 1 | Sequence 2 | Bitscore |
| SATB1 Human | SATB2 Human | 854 |
| SATB1 Shark | SATB2 Shark | 856 |
| SATB1 Human | SATB1 Shark | 1203 |
| SATB2 Human | SATB2 Shark | 1197 |
IOWs, the whole system appeared practically as it is today, before the split of cartilaginous fish and bony fish, and has retained its essential form up to now.
So, the total amount of new functional information implied by the whole system of these two proteins is about 1545 bits (considering 855 bits of common information, and 345 bits x 2 of specific information in each molecule).
An amazing amount, for a system of just two molecules, considering that 500 bits is Dembski’s Universal Probability Bound!
Let’s remember that in my previous post, quoted above, I showed that the informational jump from pre-vertebrates to vertebrates is more than 1.7 million bits. That’s a very big number, but big numbers sometimes are not easily digested. So, I believe that seeing that just two important molecules can contribute for almost 1500 bits can help us understand what we are really seeing here.
Moreover, it’s certainly not a case that those two molecules seem to be fundamental in two very particular fields:
a) The adaptive immune system
b) The nervous system
if we consider that those are exactly the two most relevant developments in vertebrates.
And, as a final note, please consider that these are very complex master regulators, which interact with tens of other complex proteins to effect their functions. The whole system is certainly much more irreducibly complex than we can imagine.
But still, just the analysis of these two sister proteins is more than enough to demonstrate that the neo Darwinian paradigm is completely inappropriate to explain what we can see in the proteome and in its natural history. And this is only one example among thousands.
So, I want to conclude repeating again this strong and very convinced statement:
The observed facts described here cannot in any way be explained by any neo-darwinian model. Absolutely not. They are extremely strong evidence for a design inference.